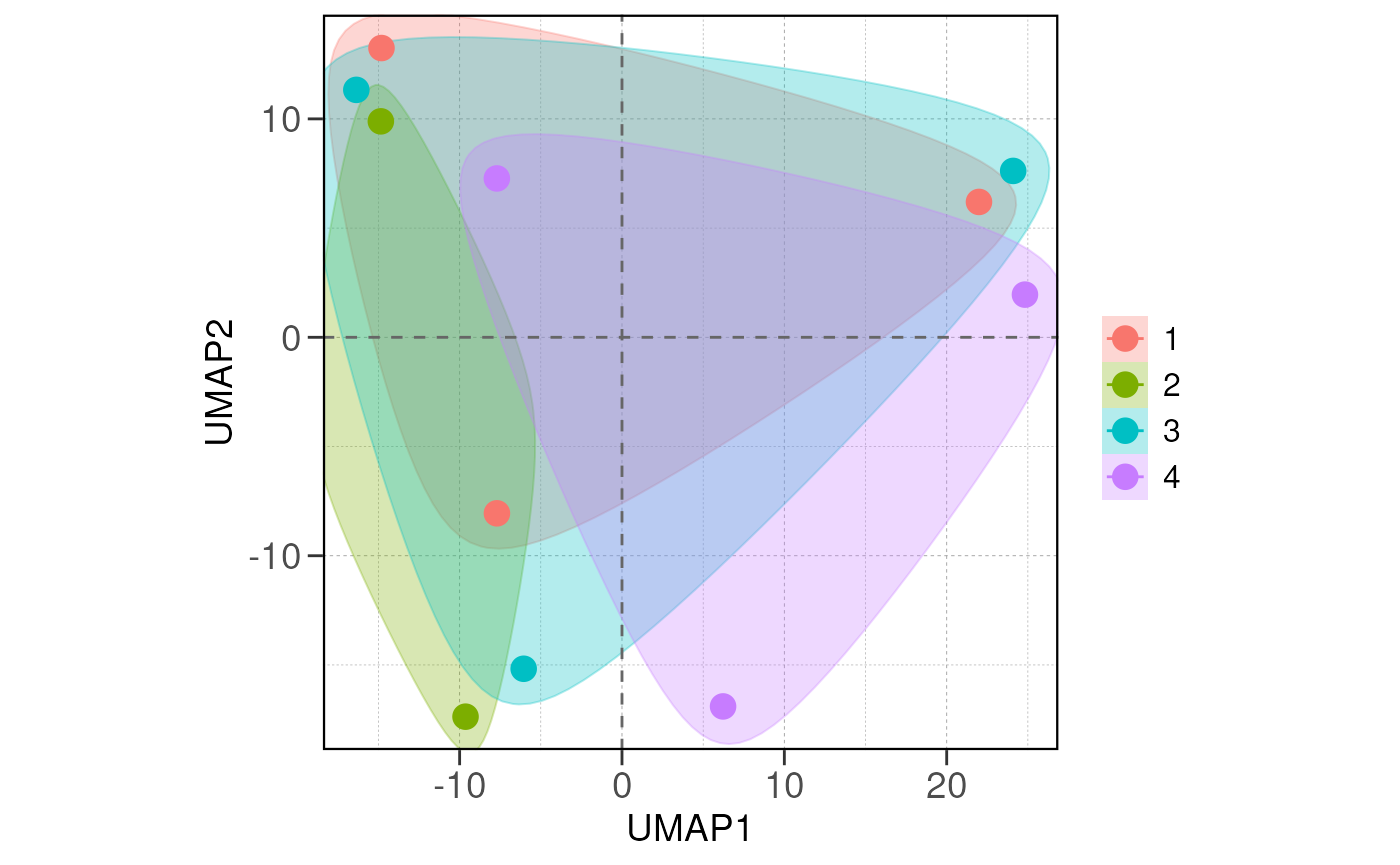
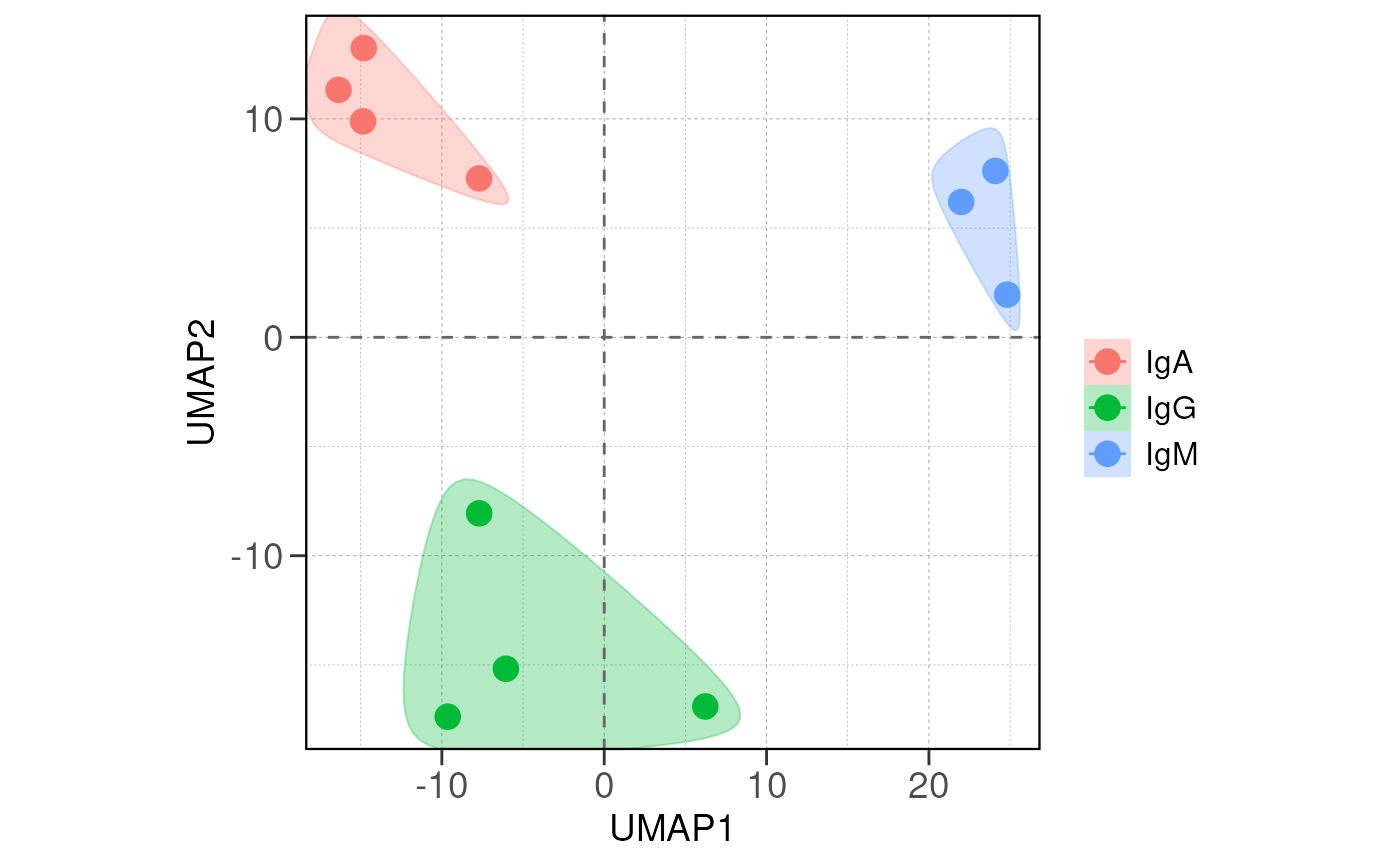
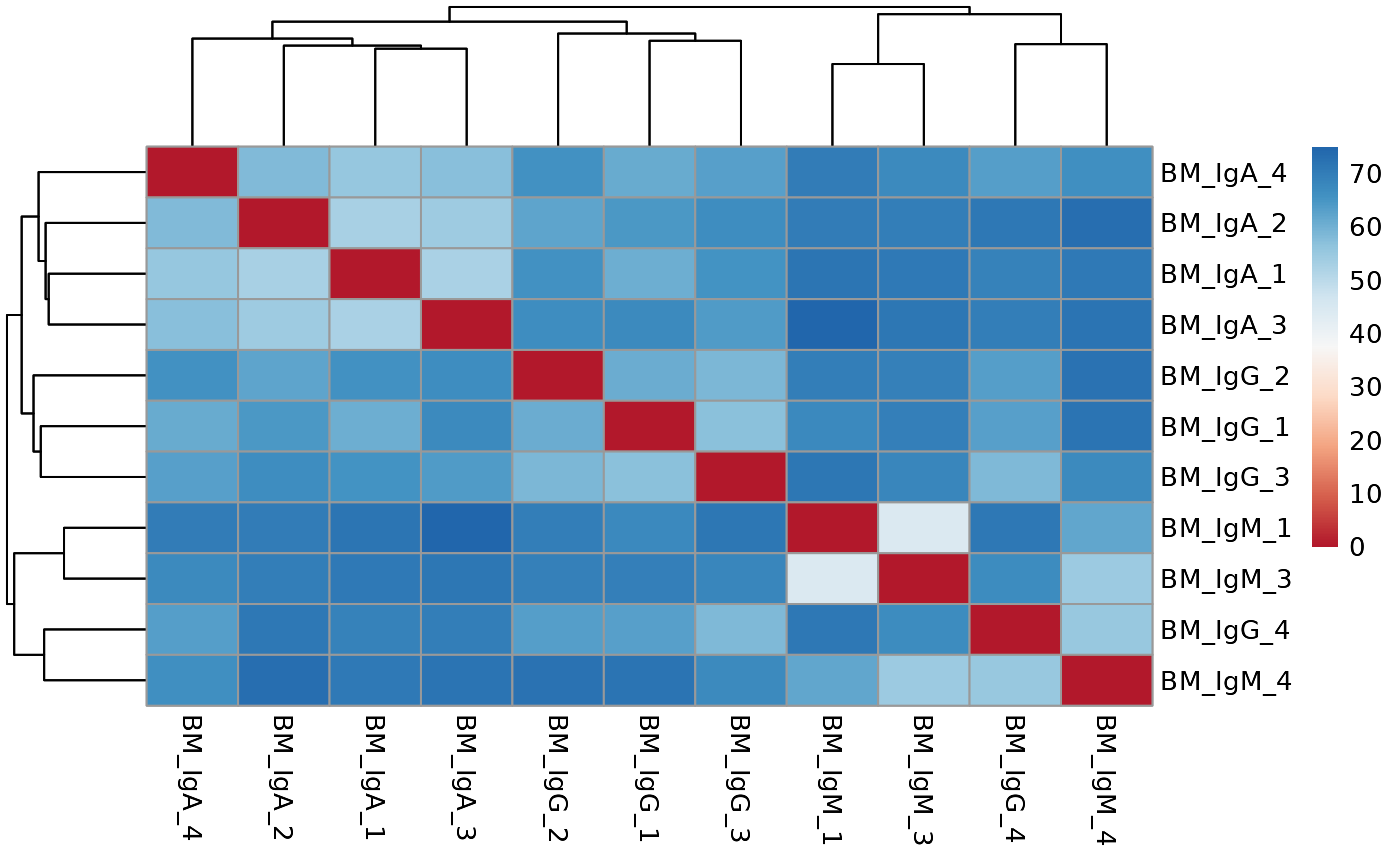
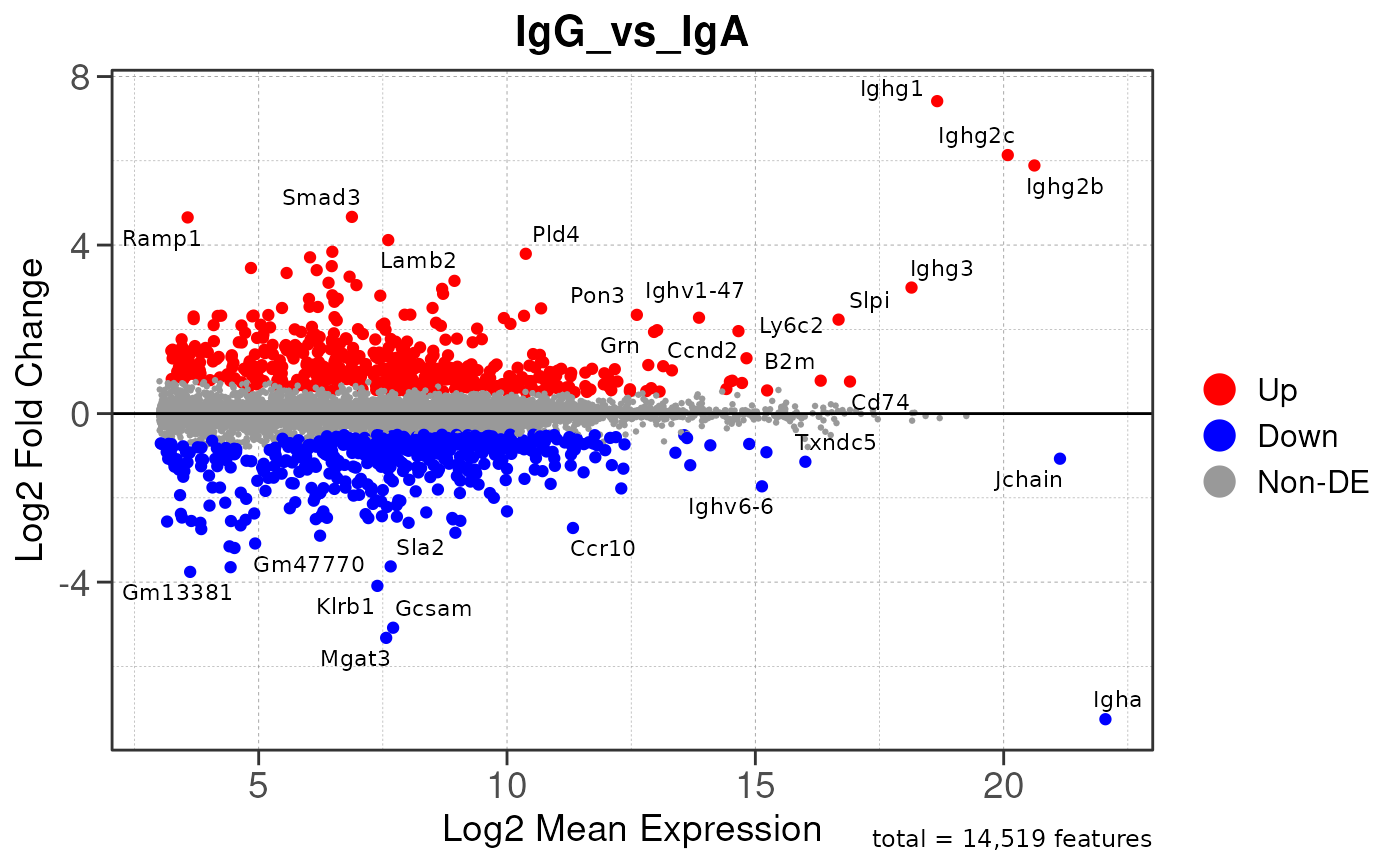
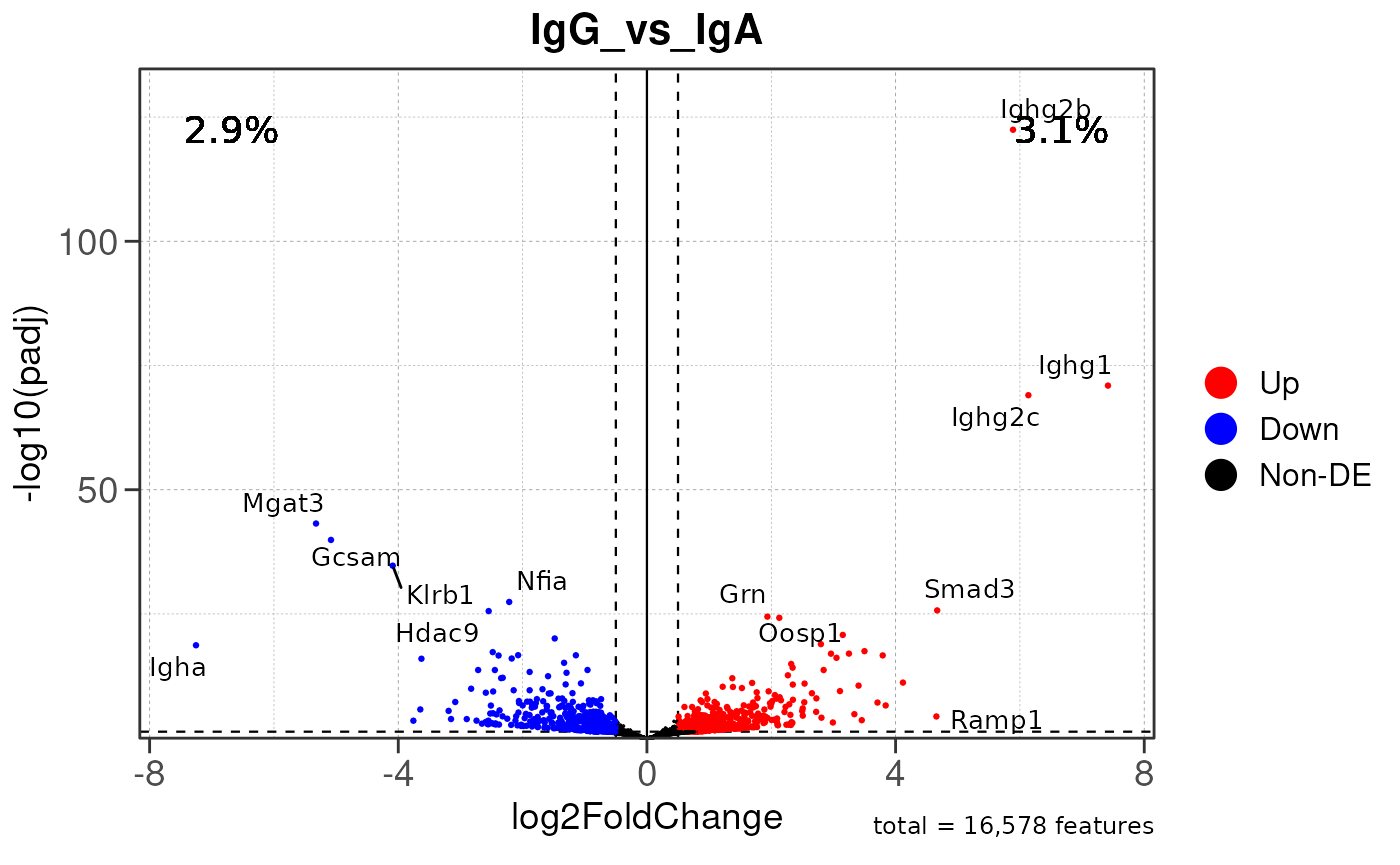
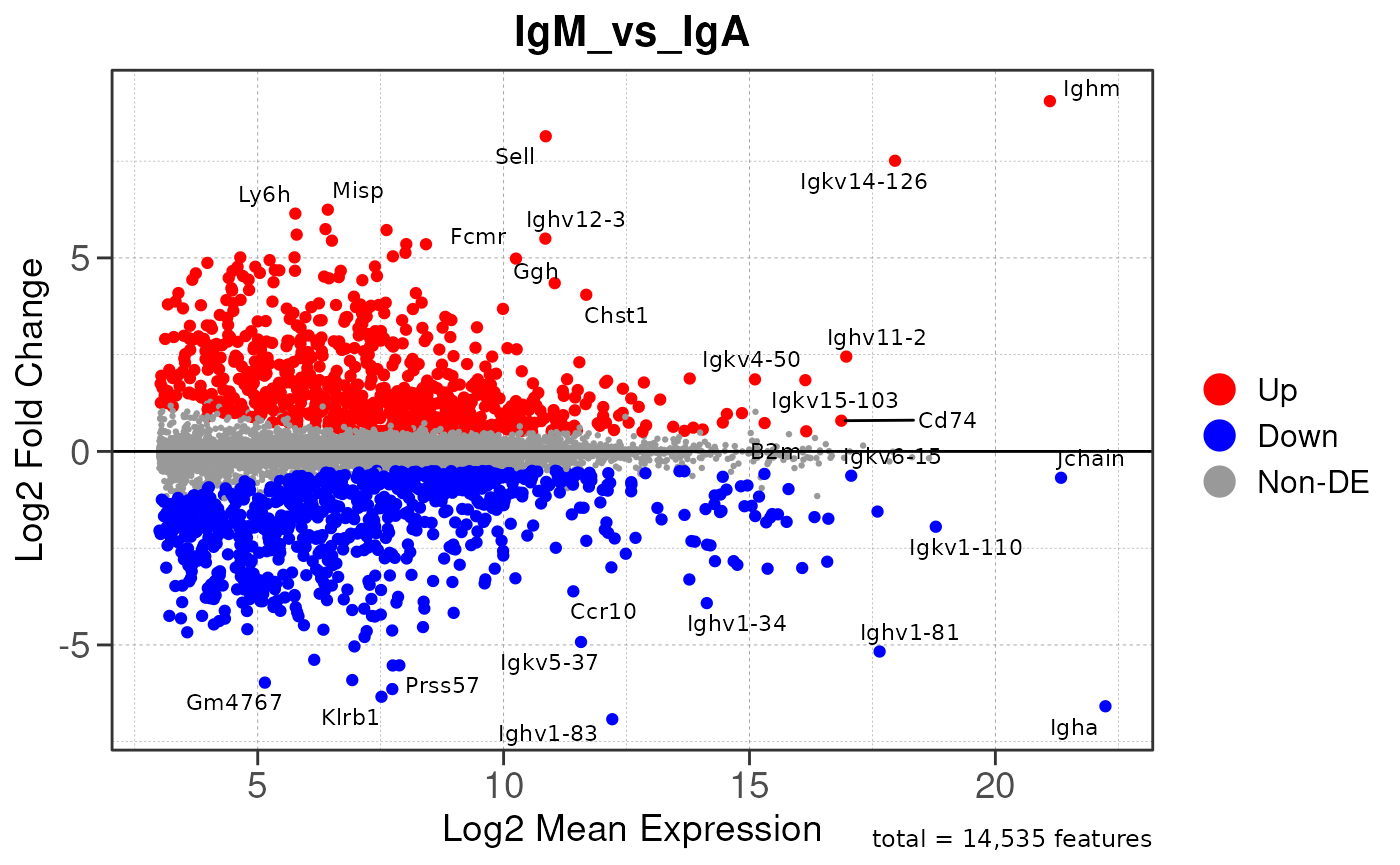
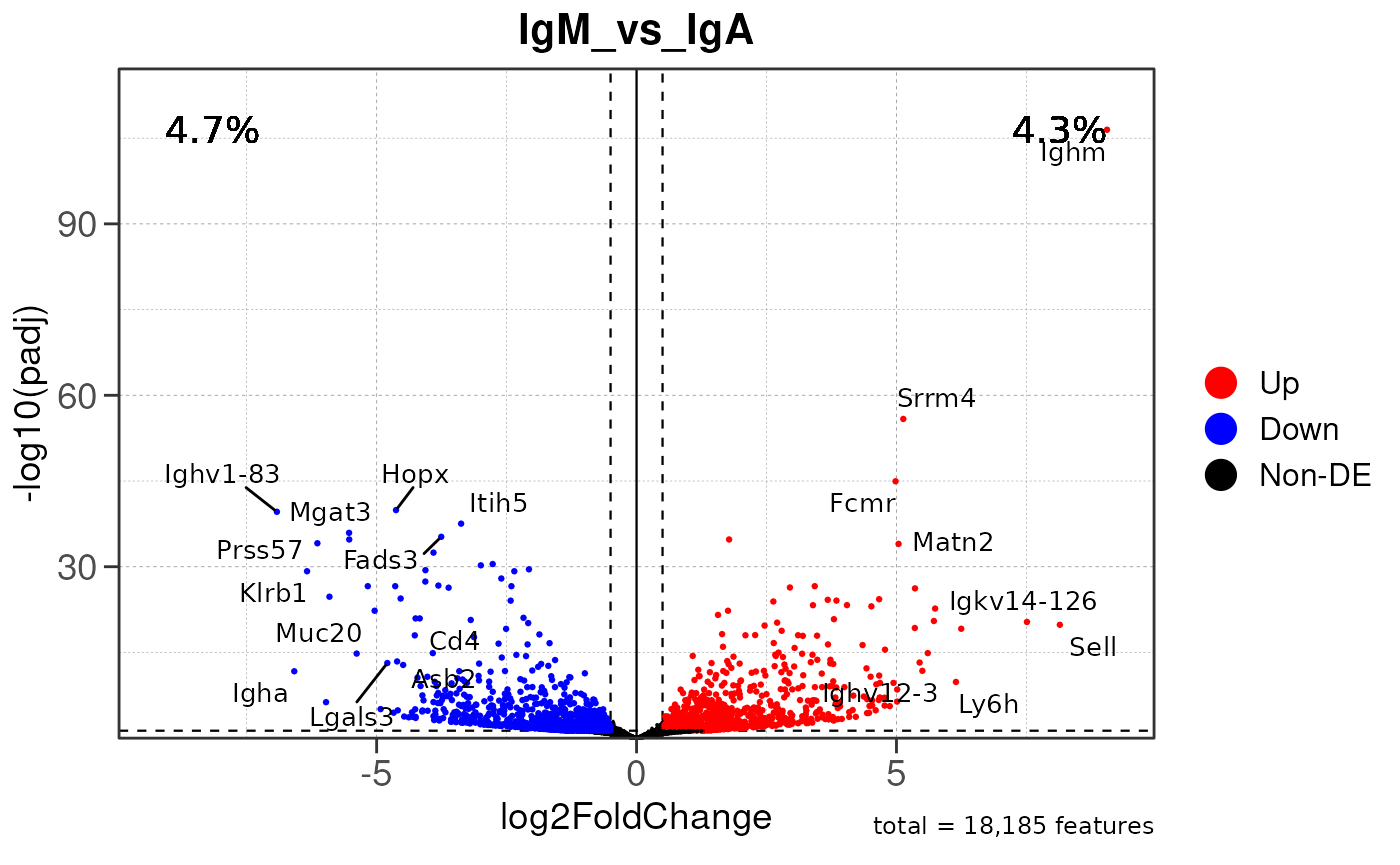
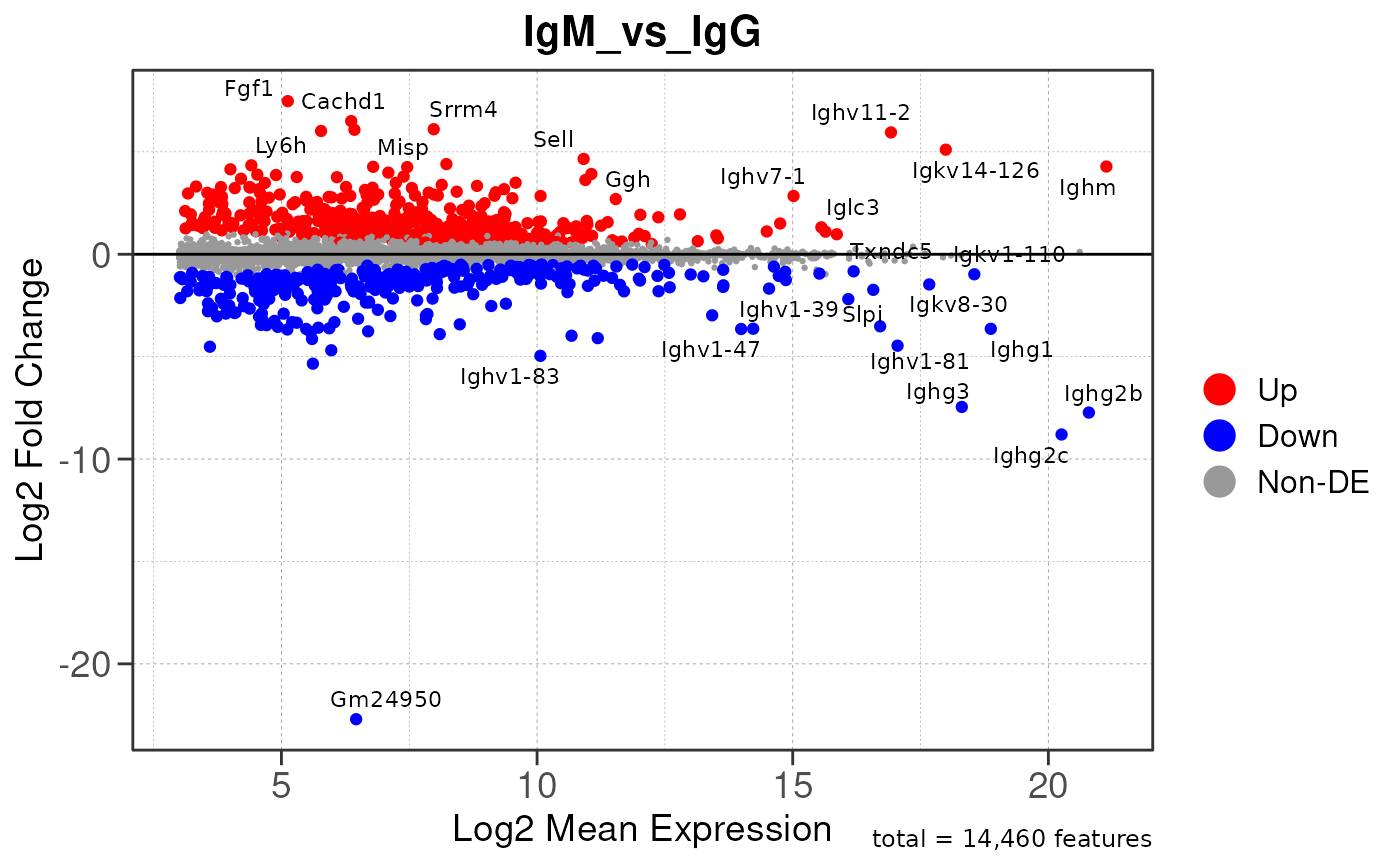
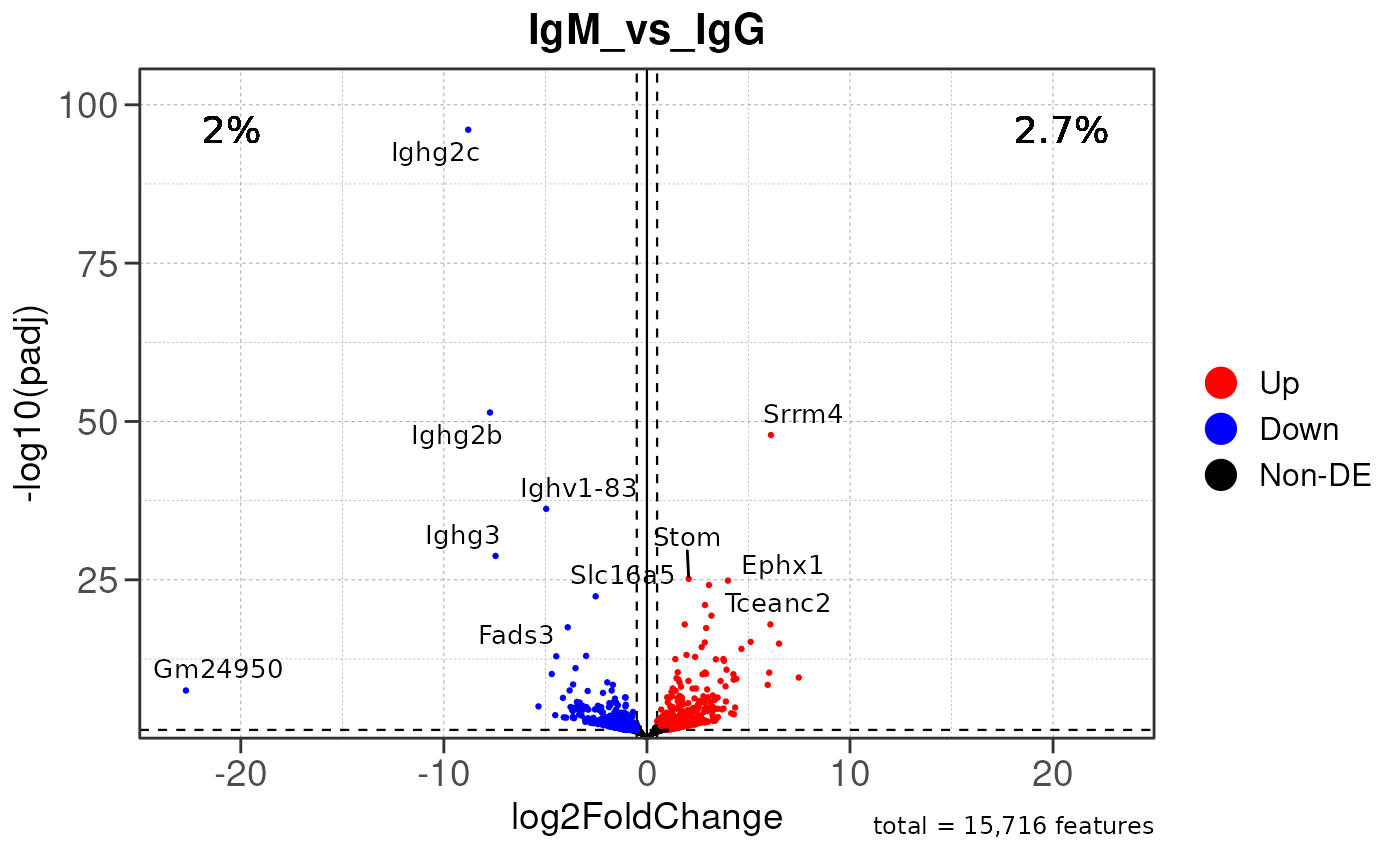
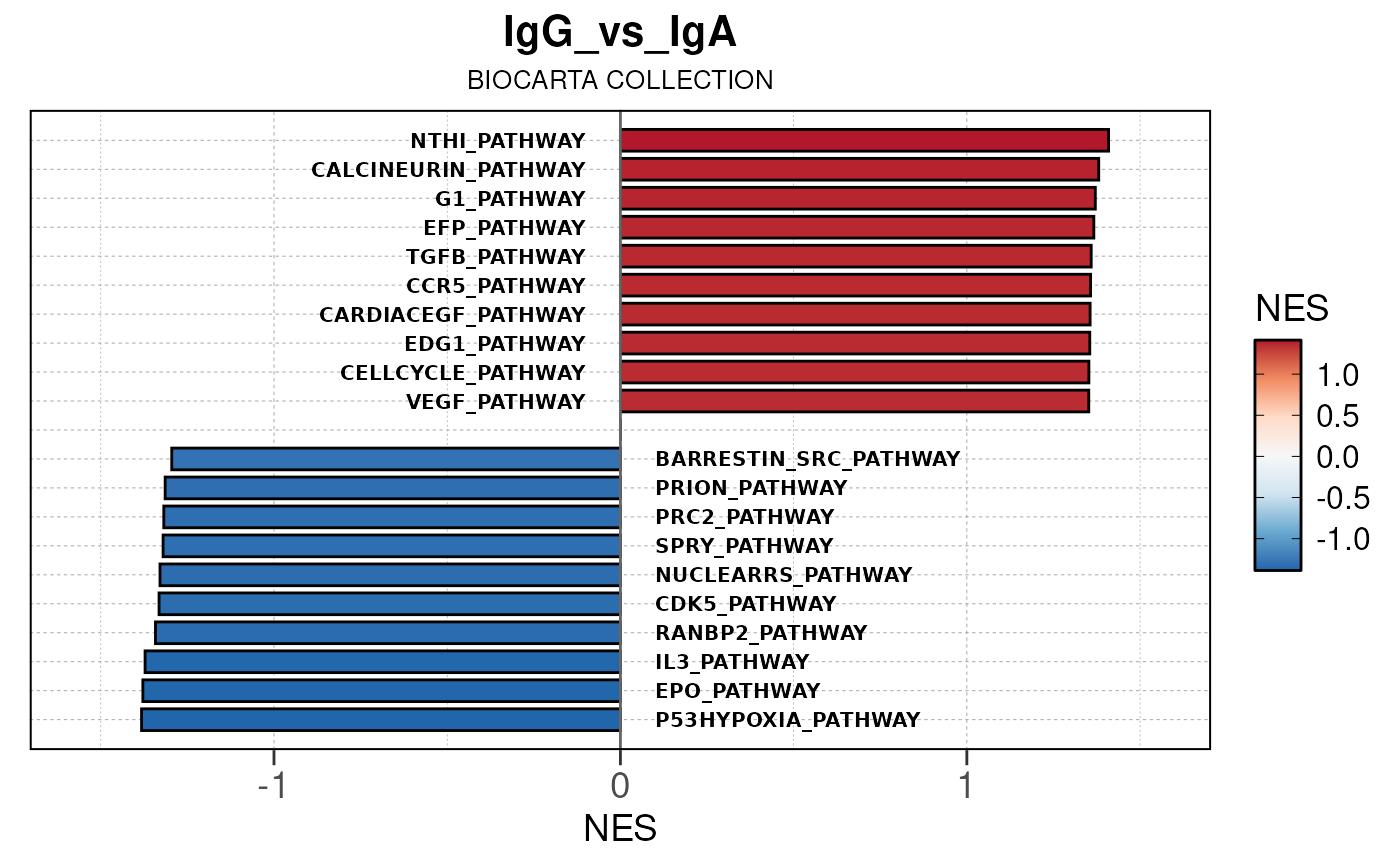
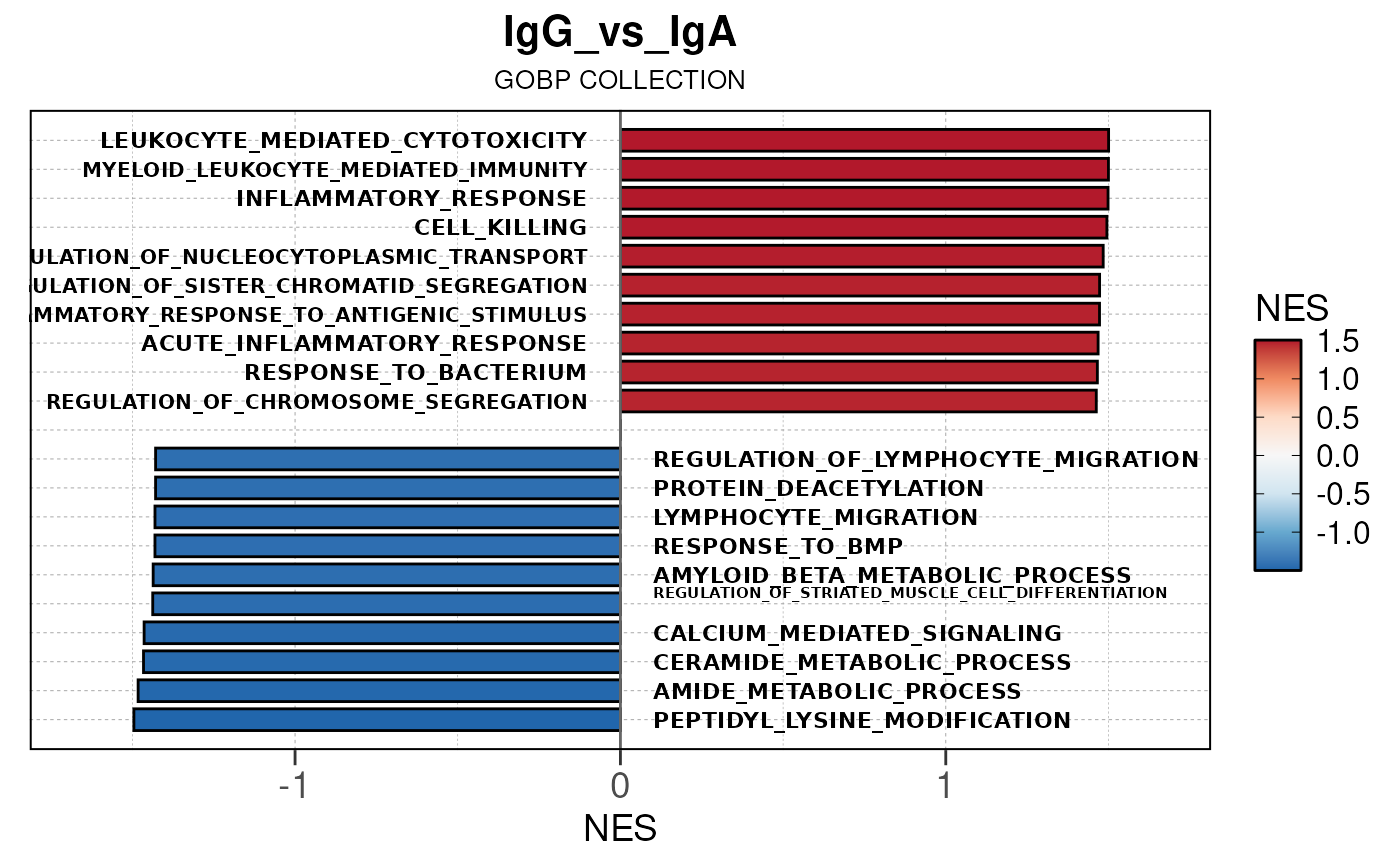
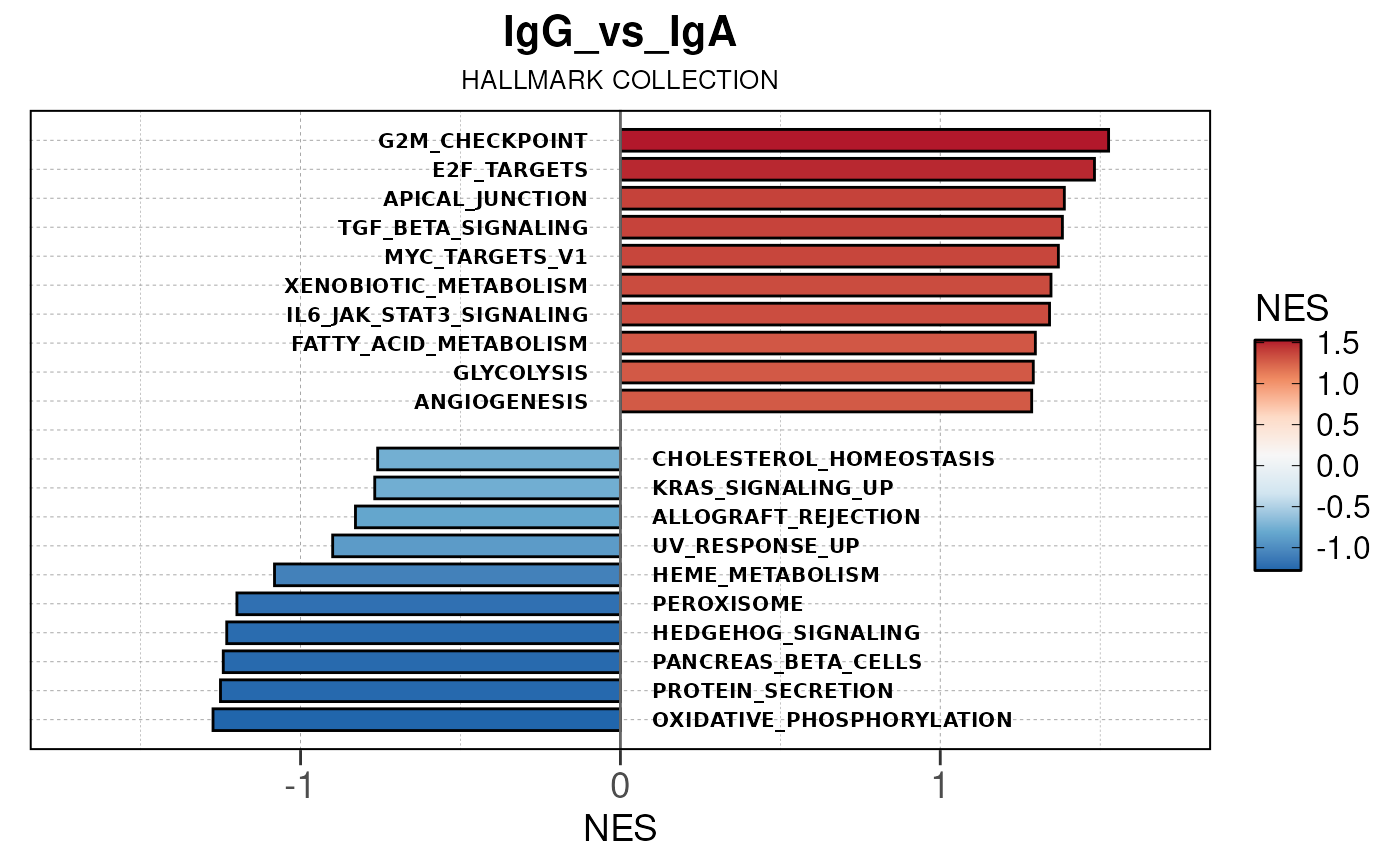
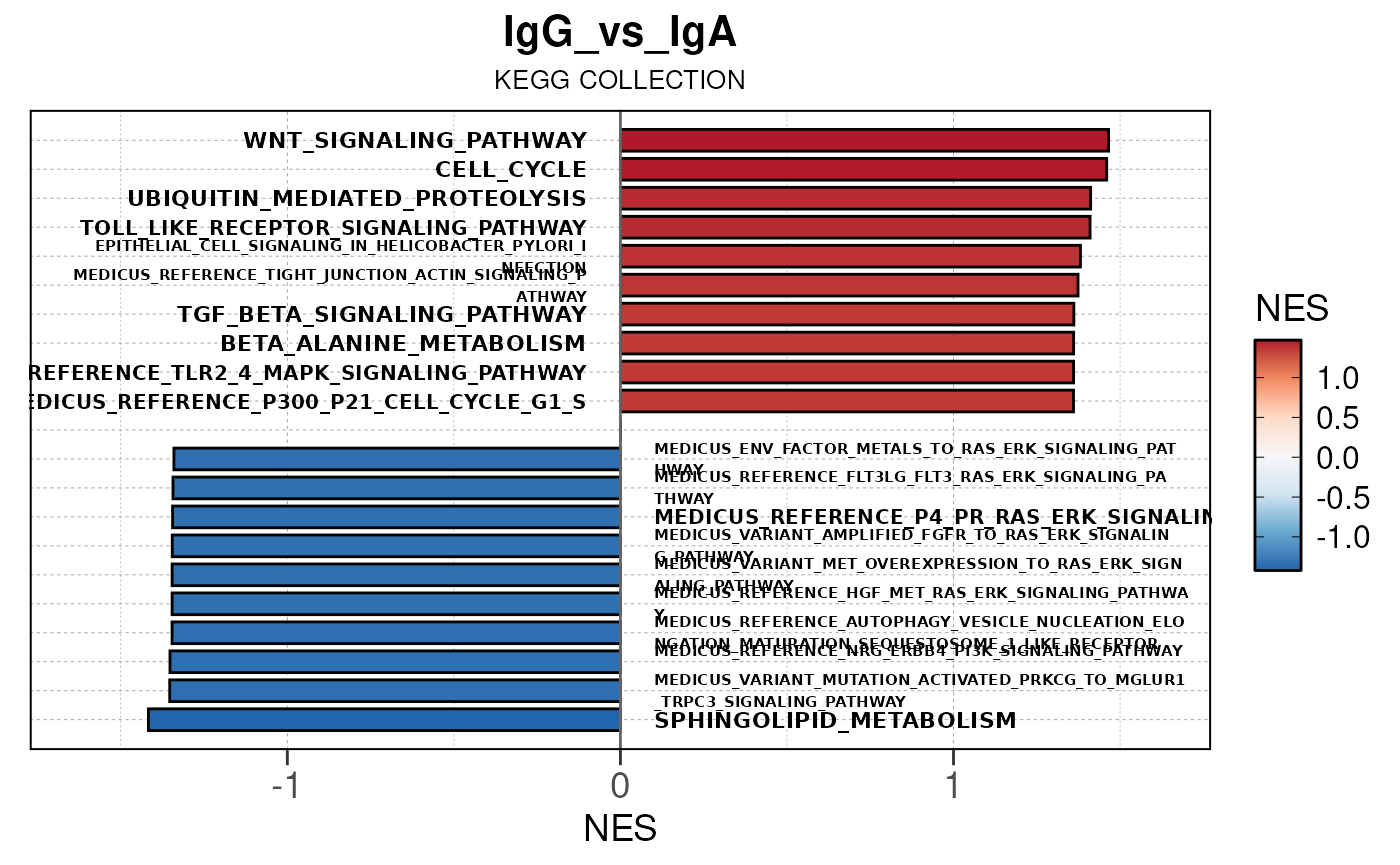
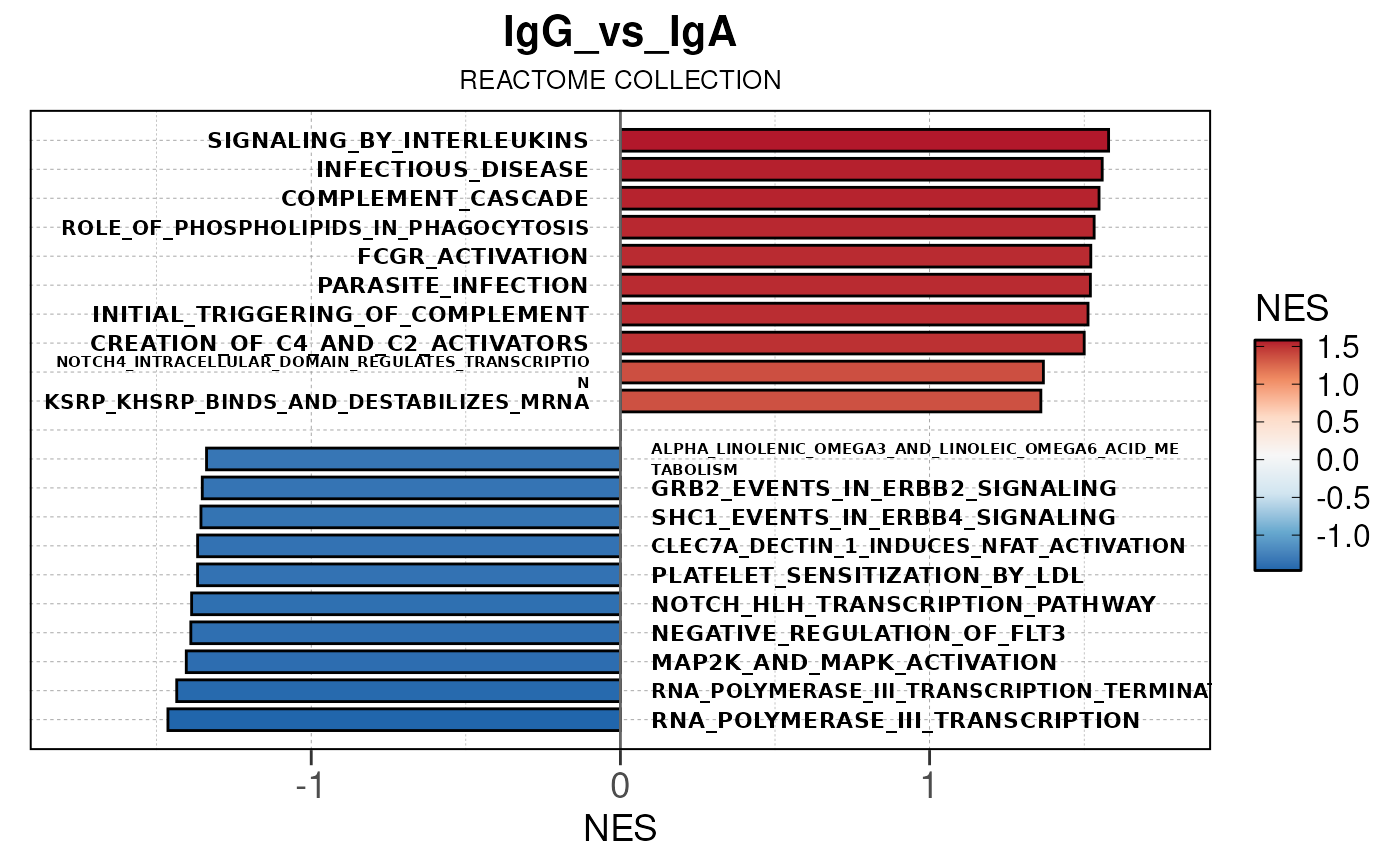
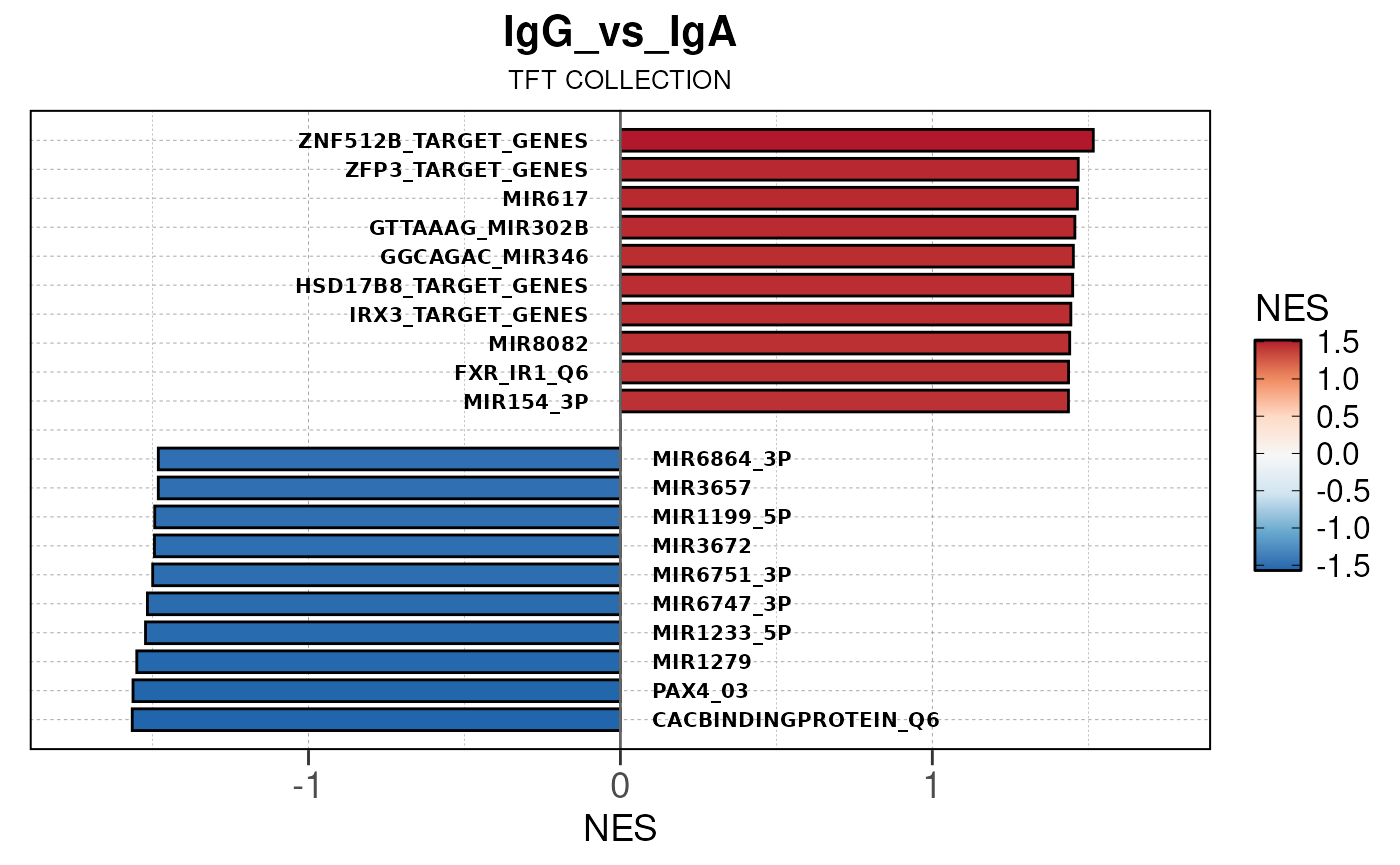
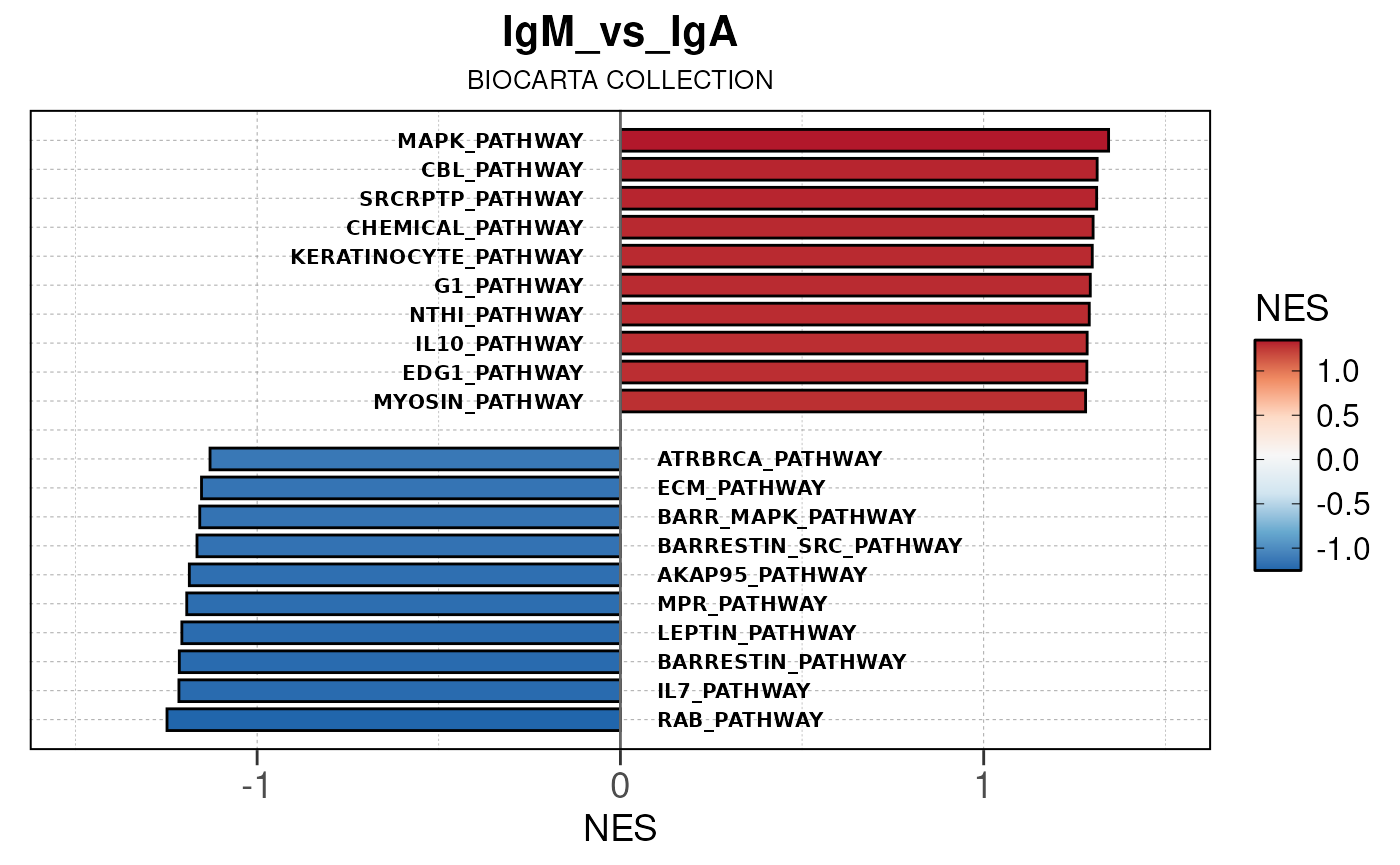
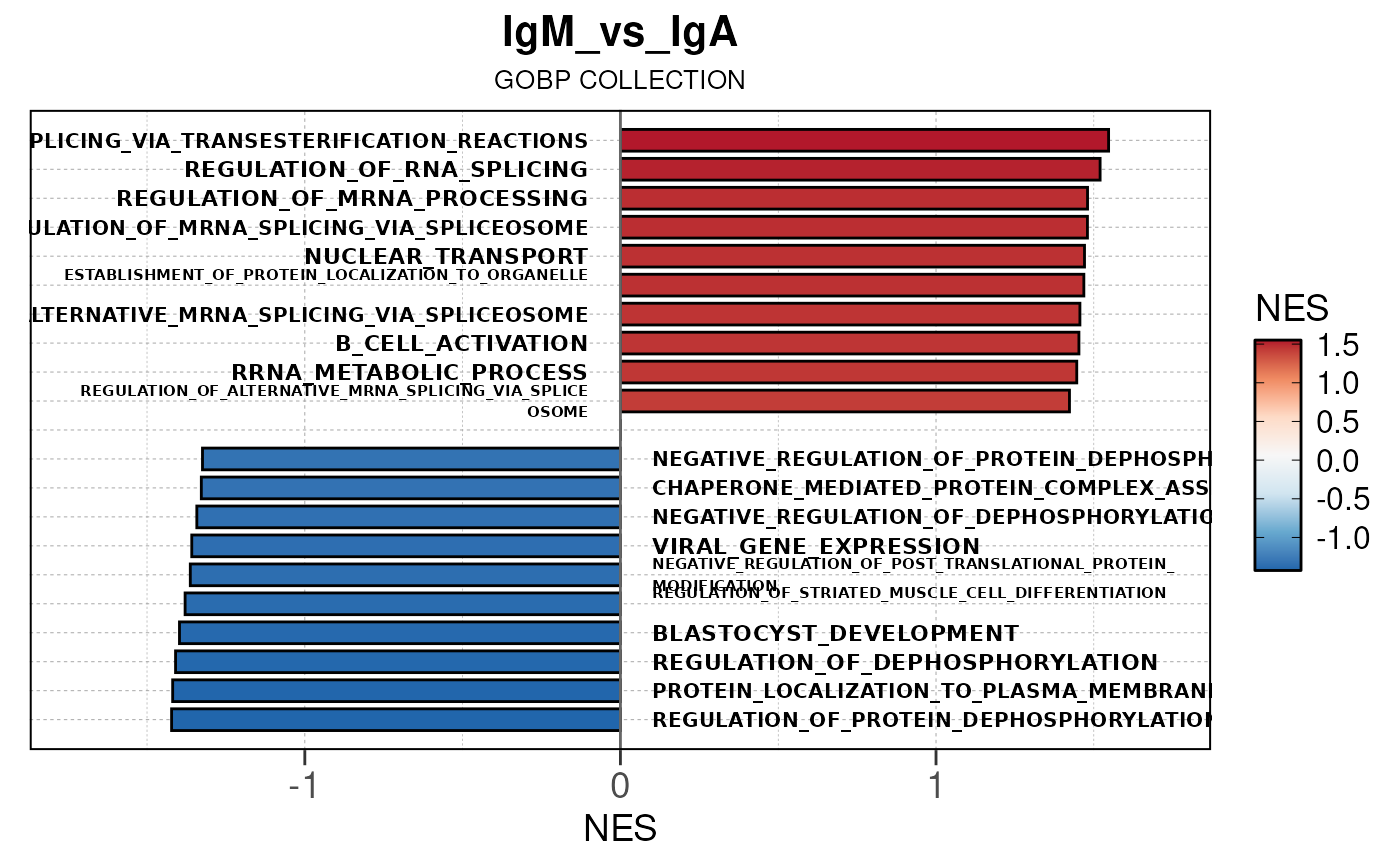
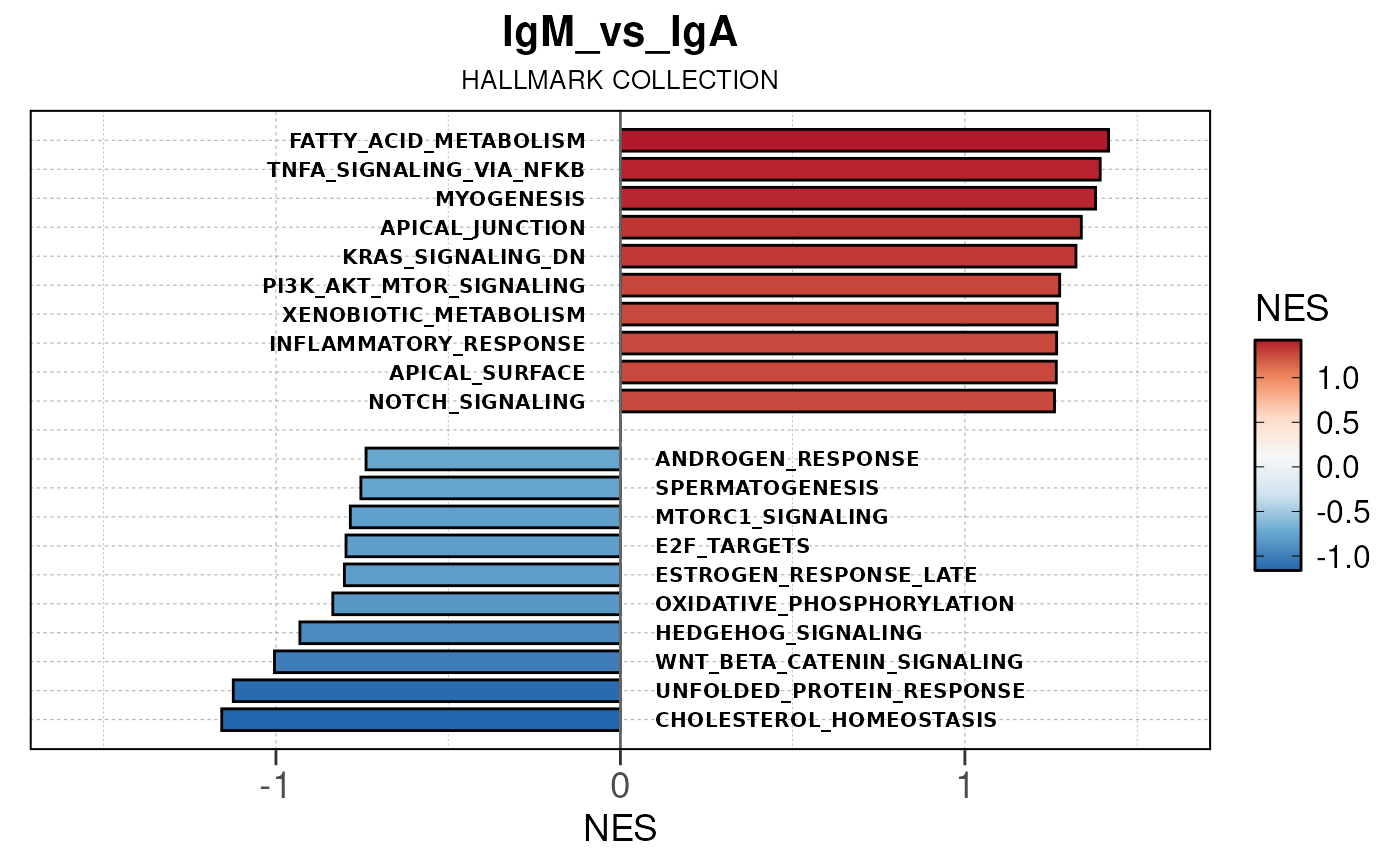
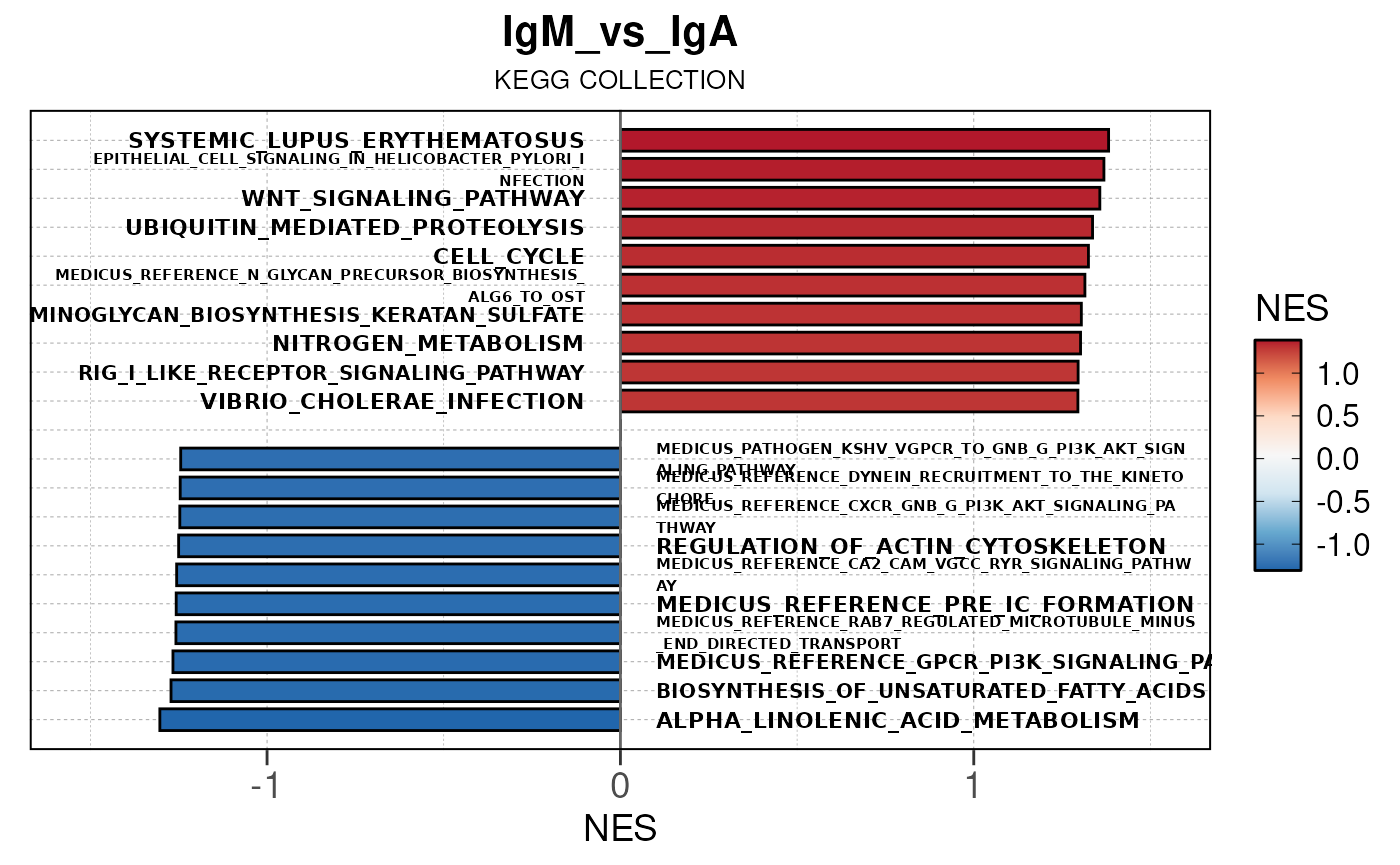
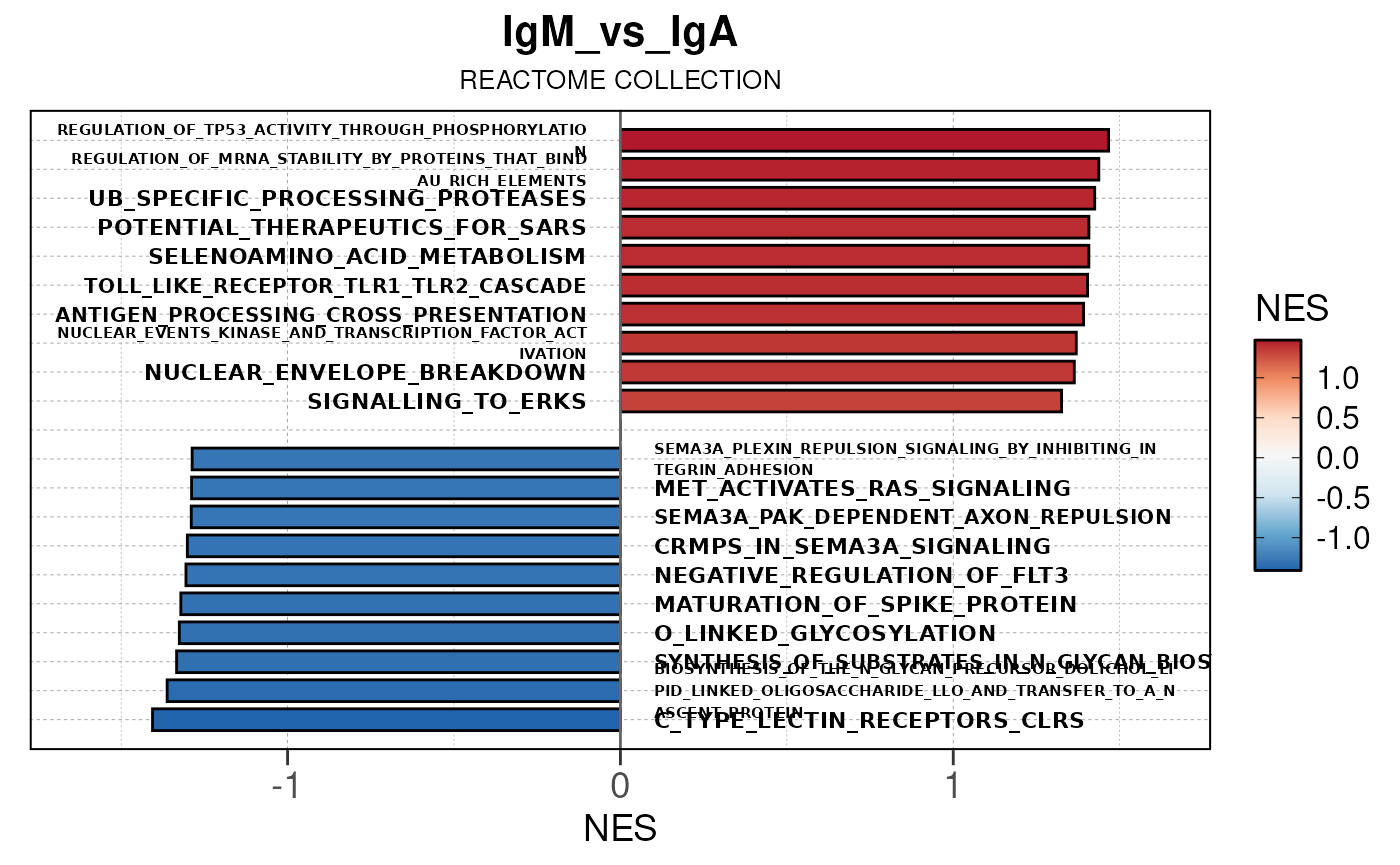
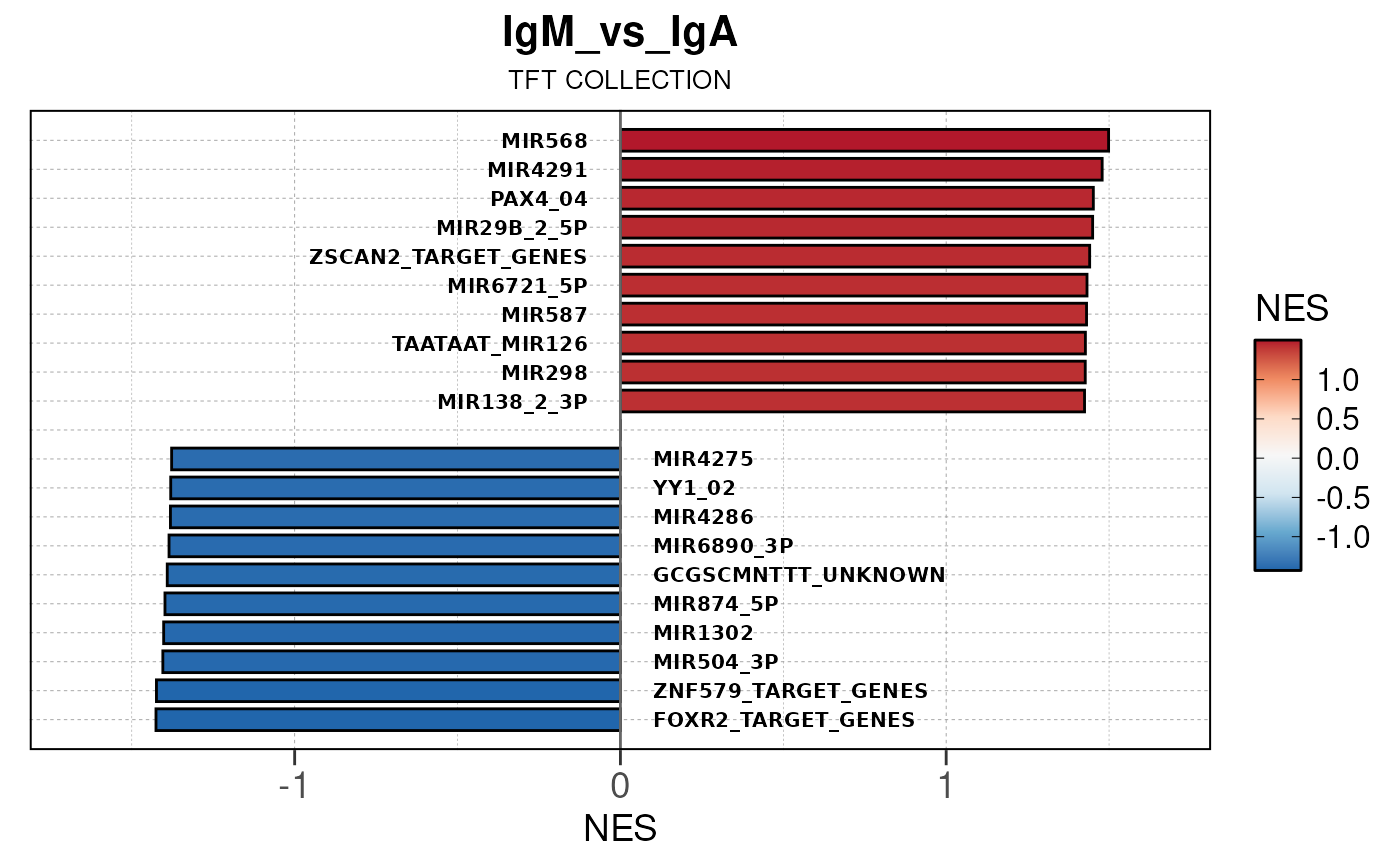
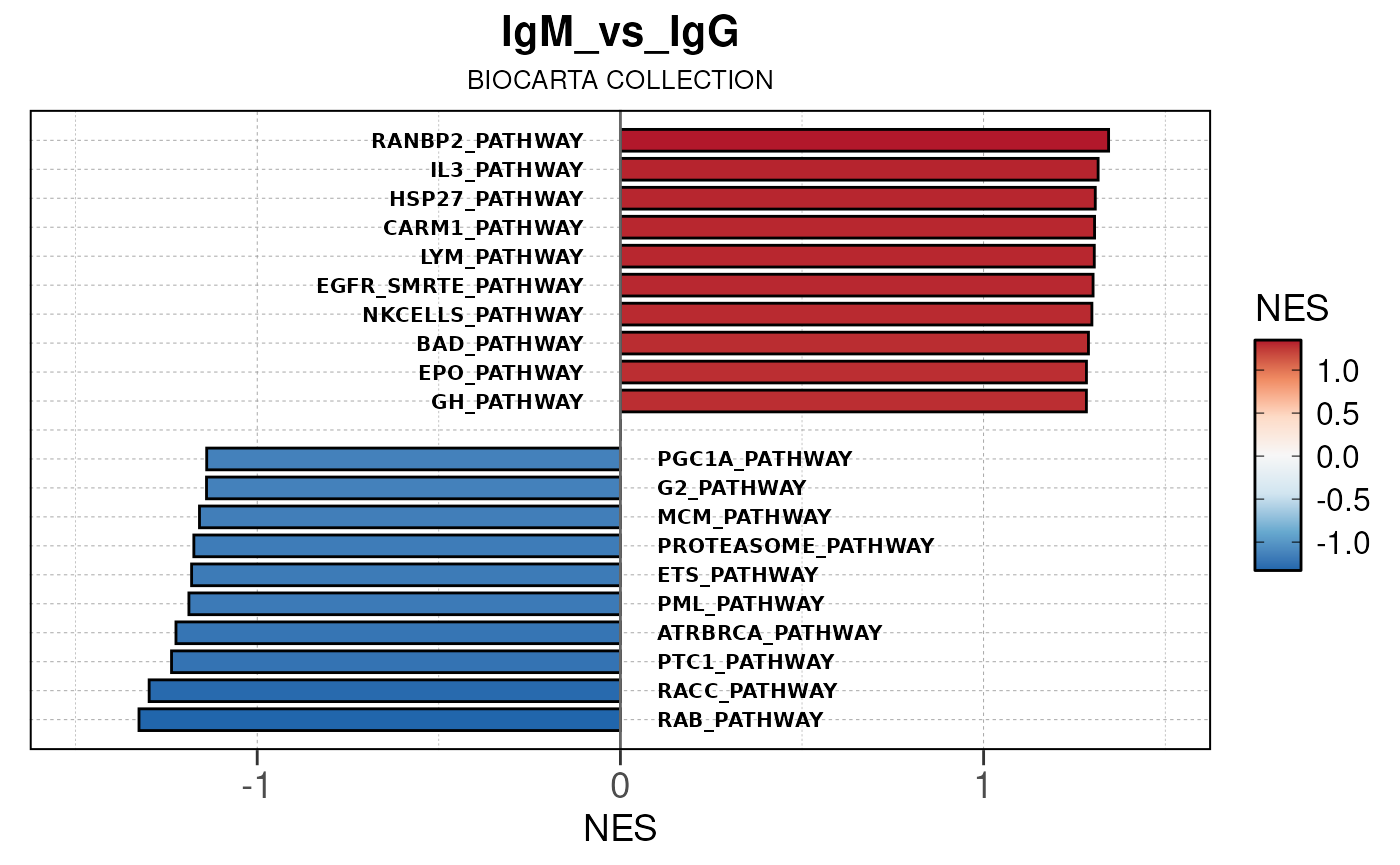
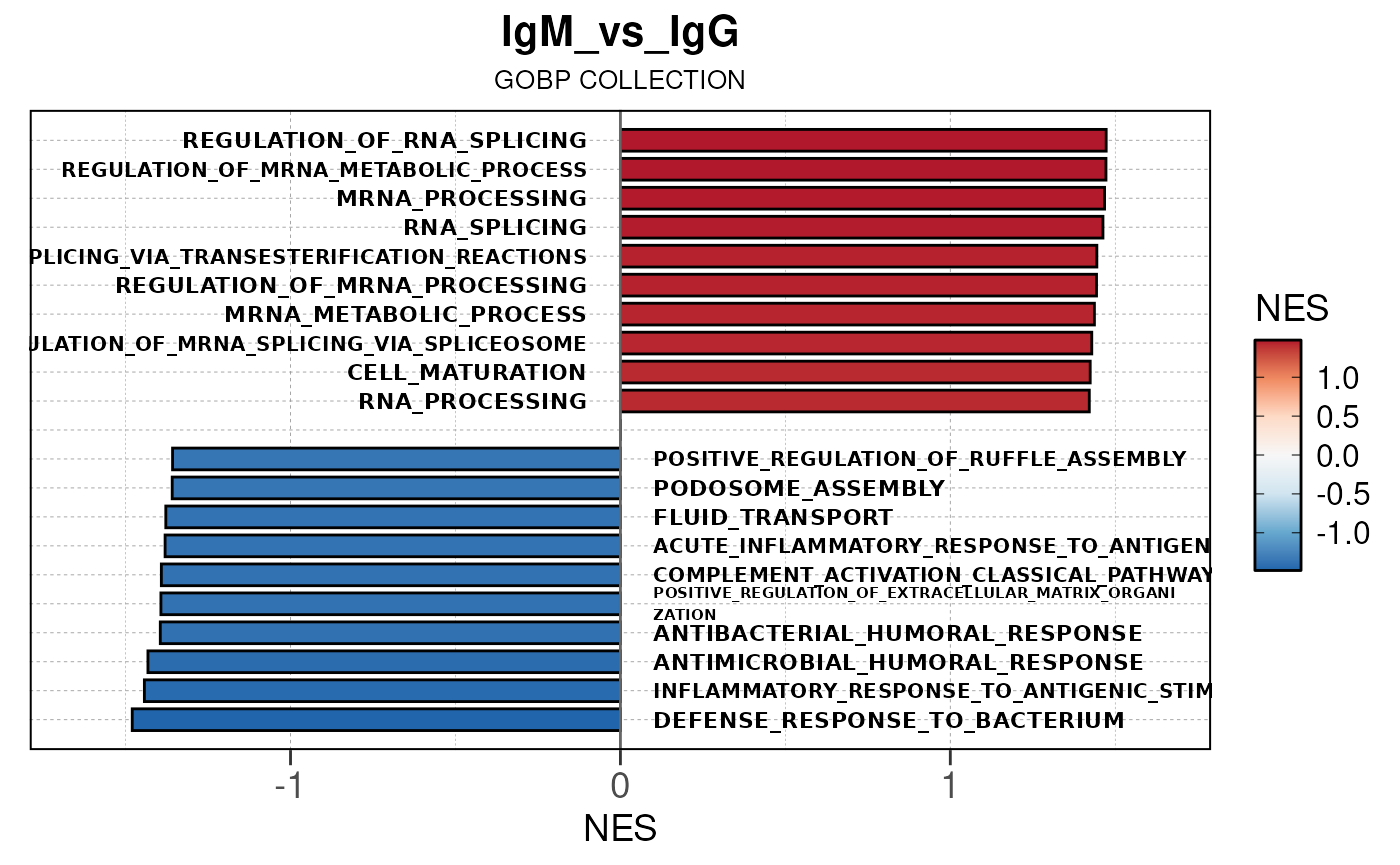
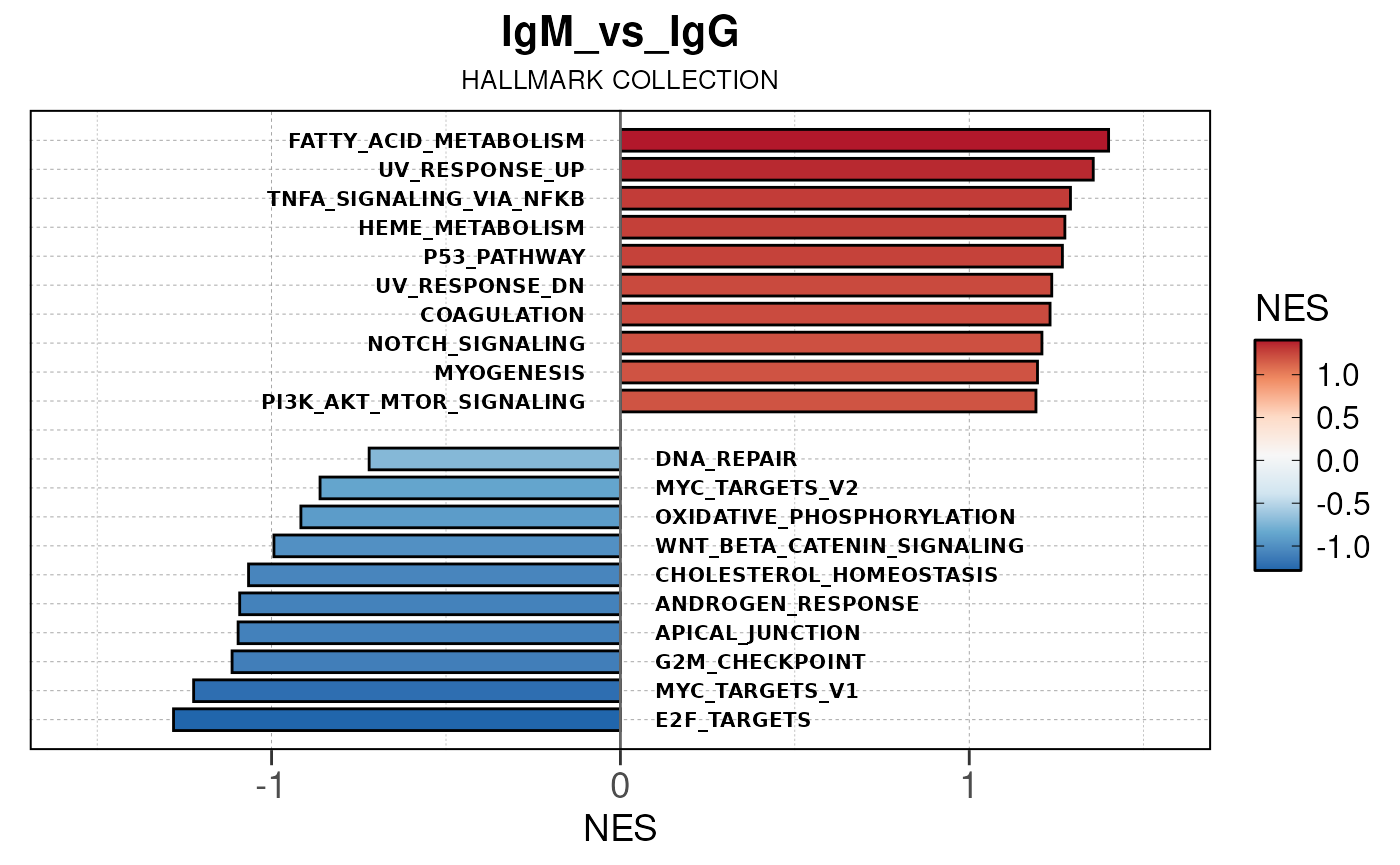
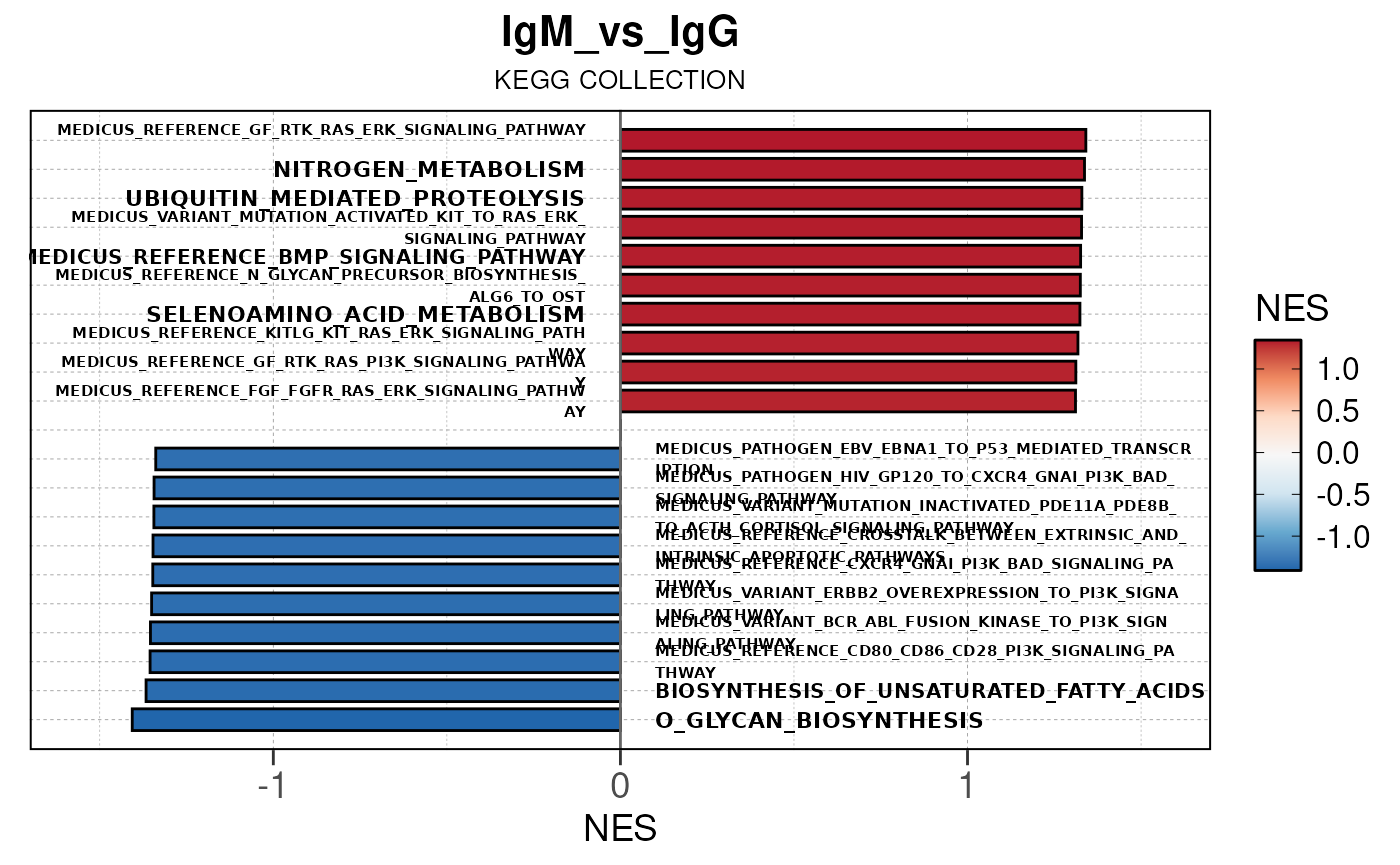
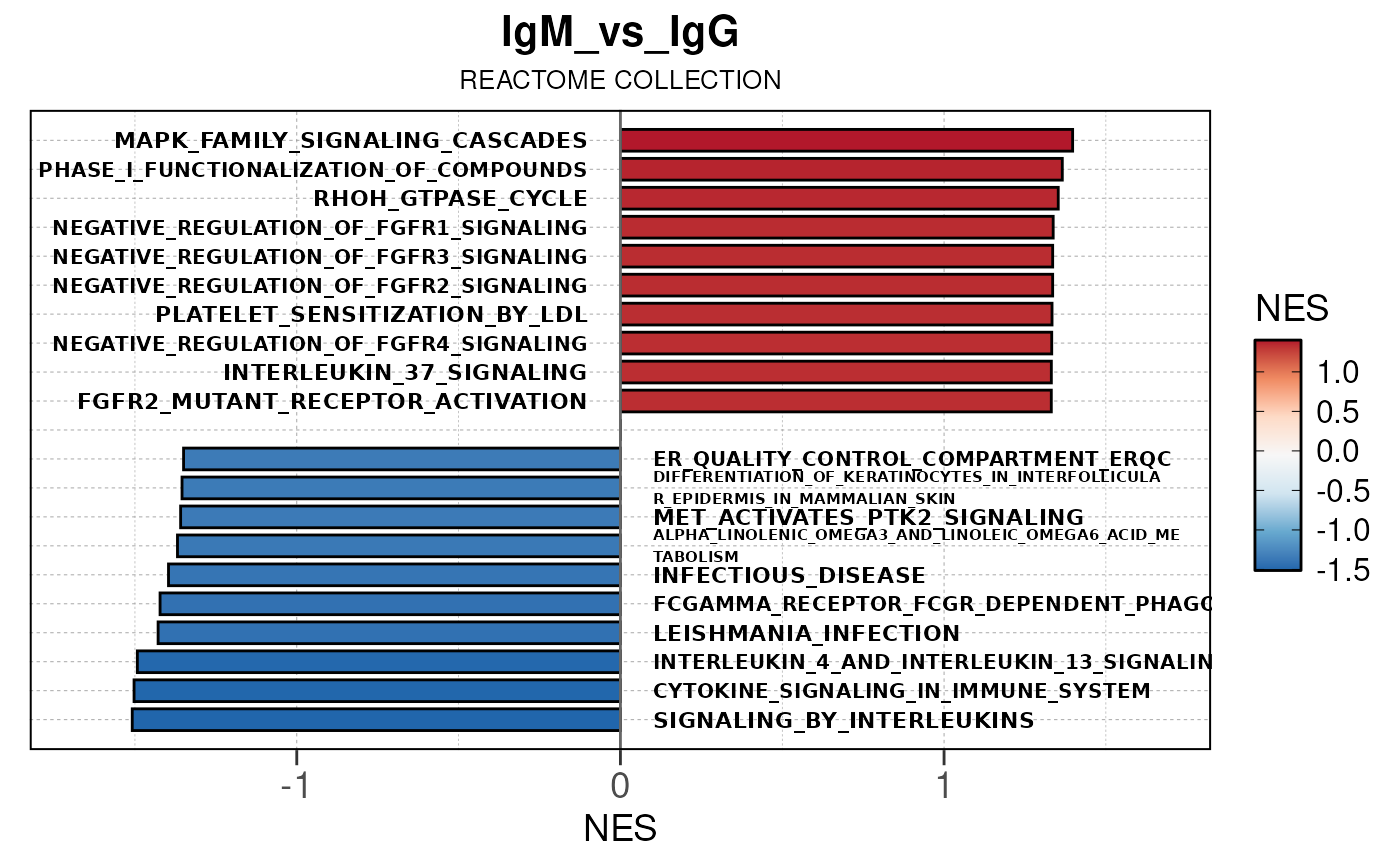
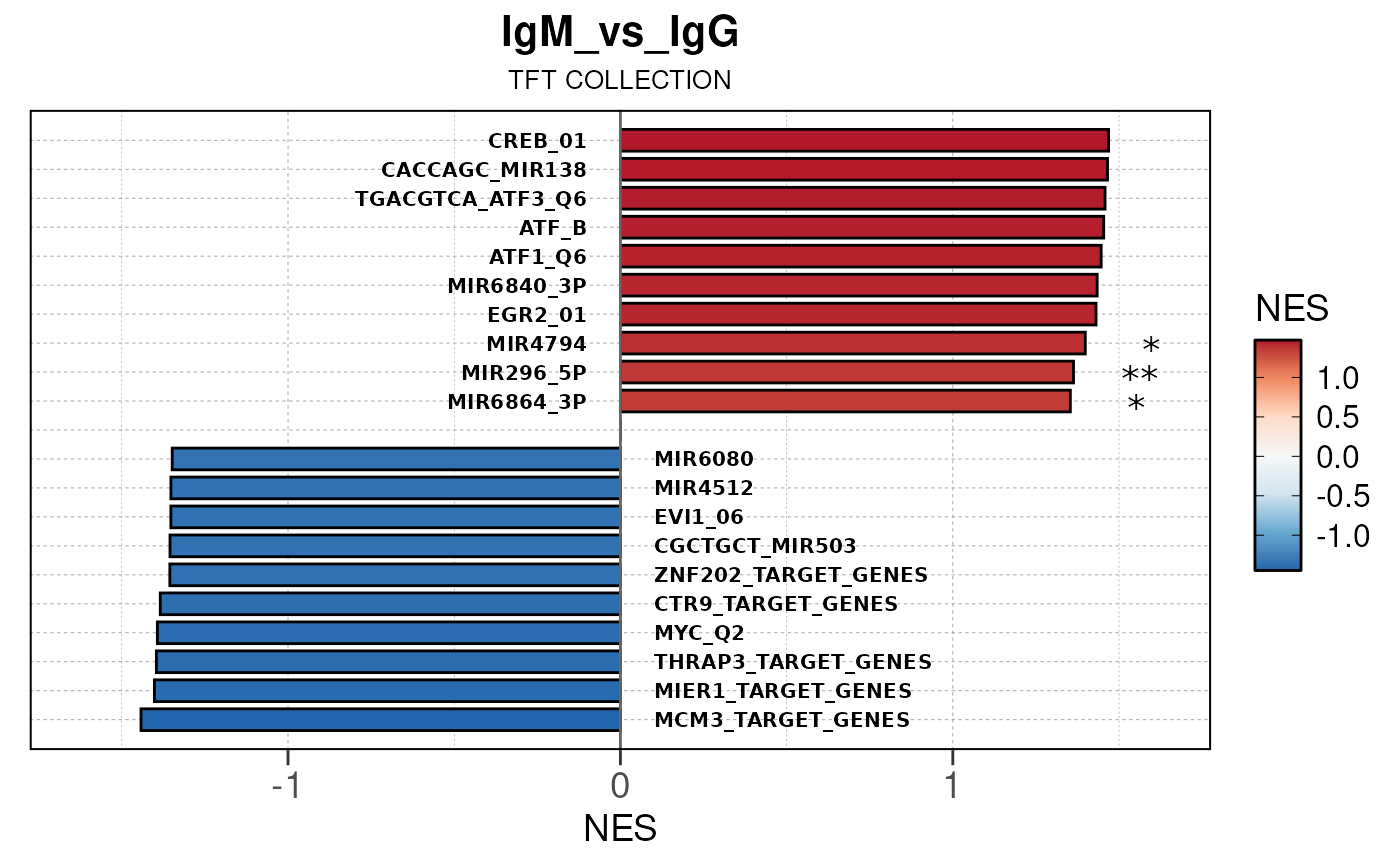
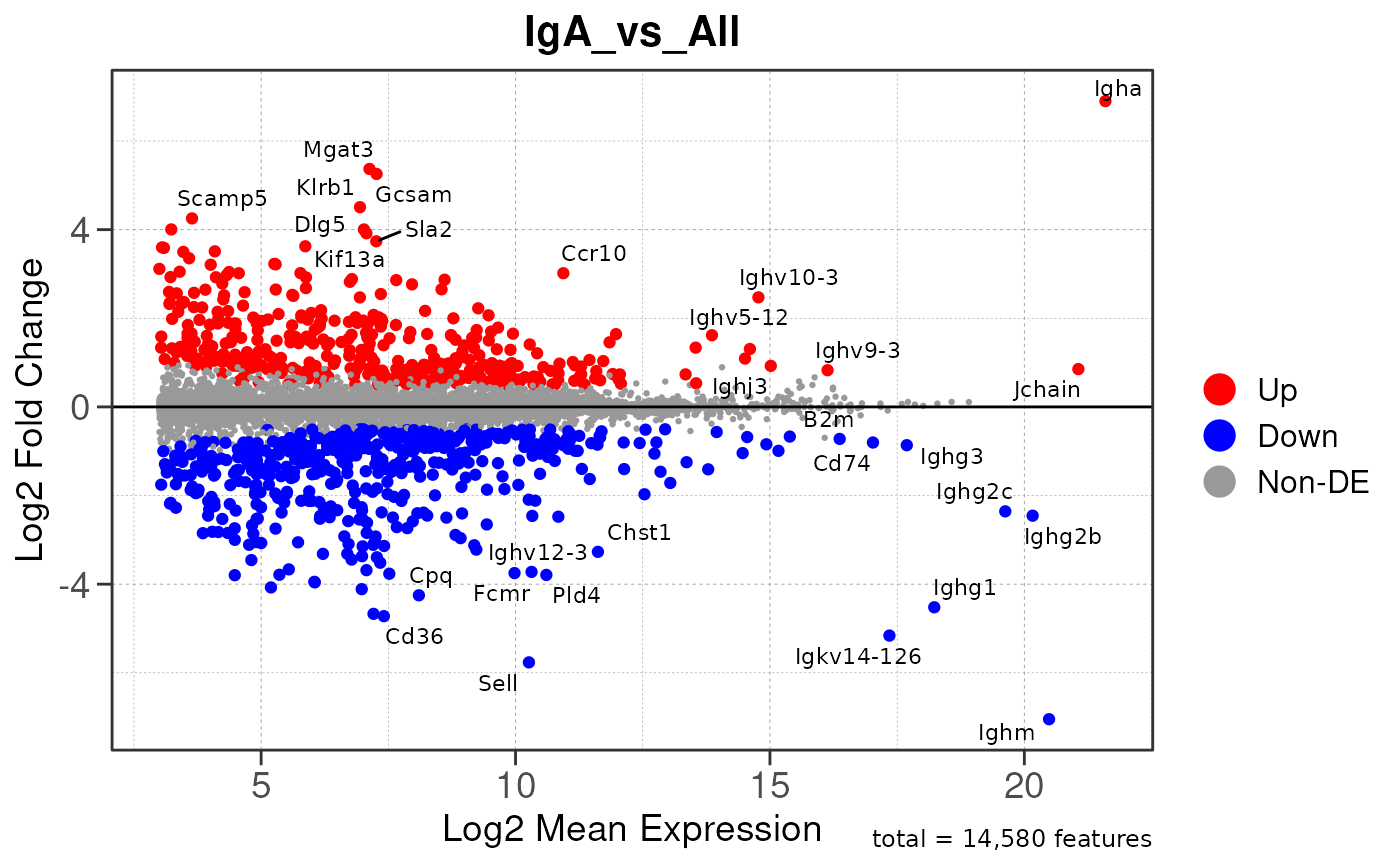
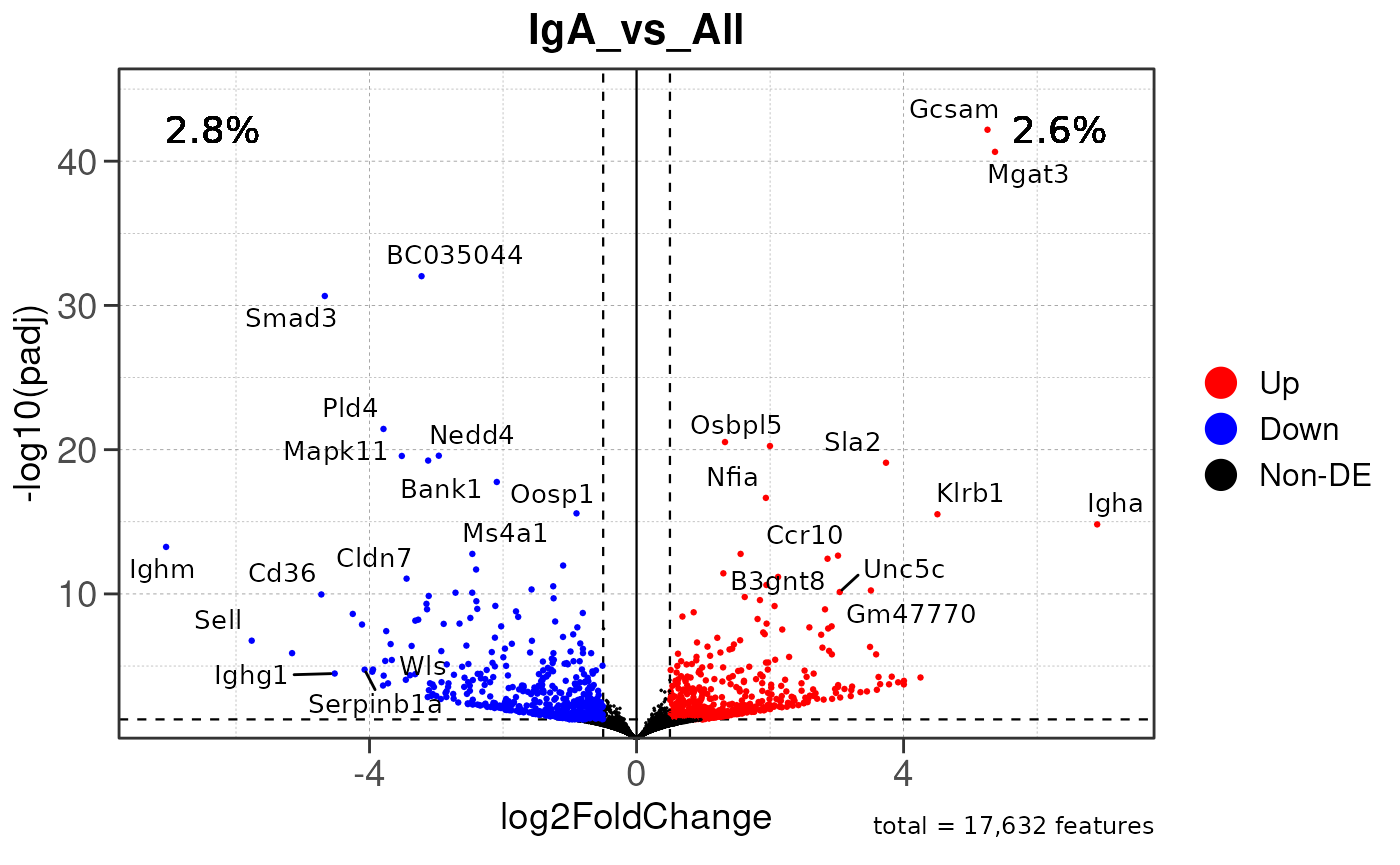
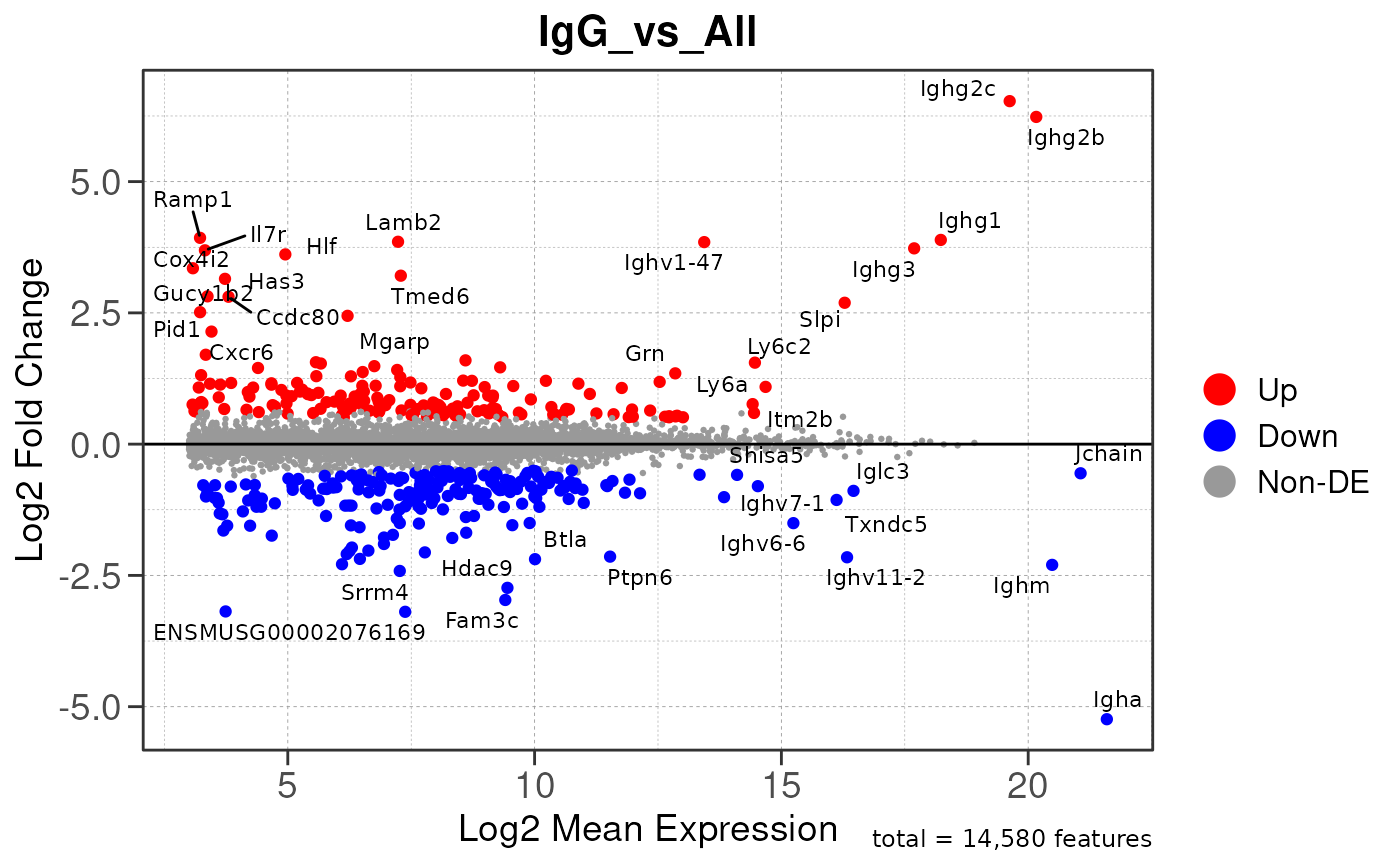
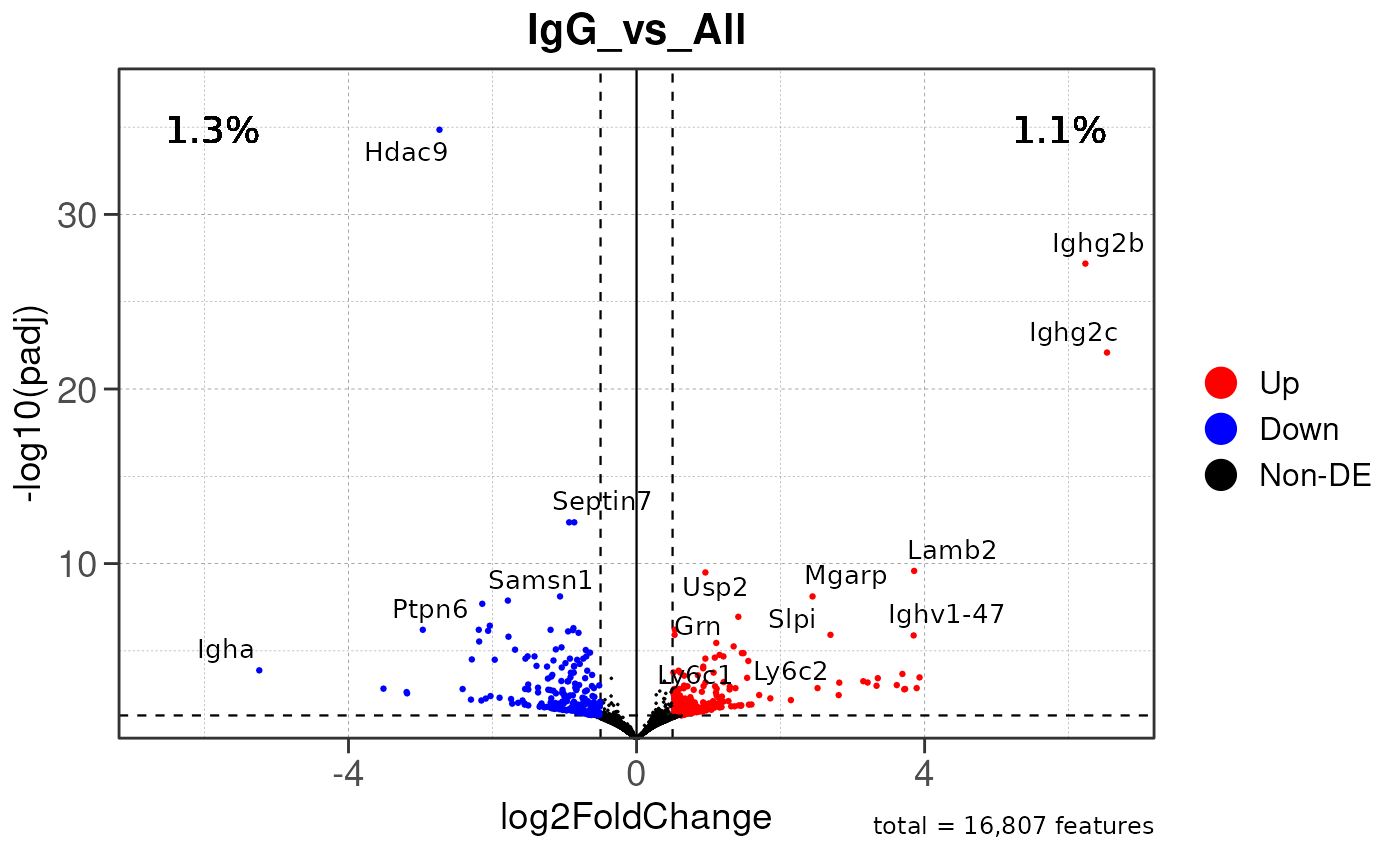
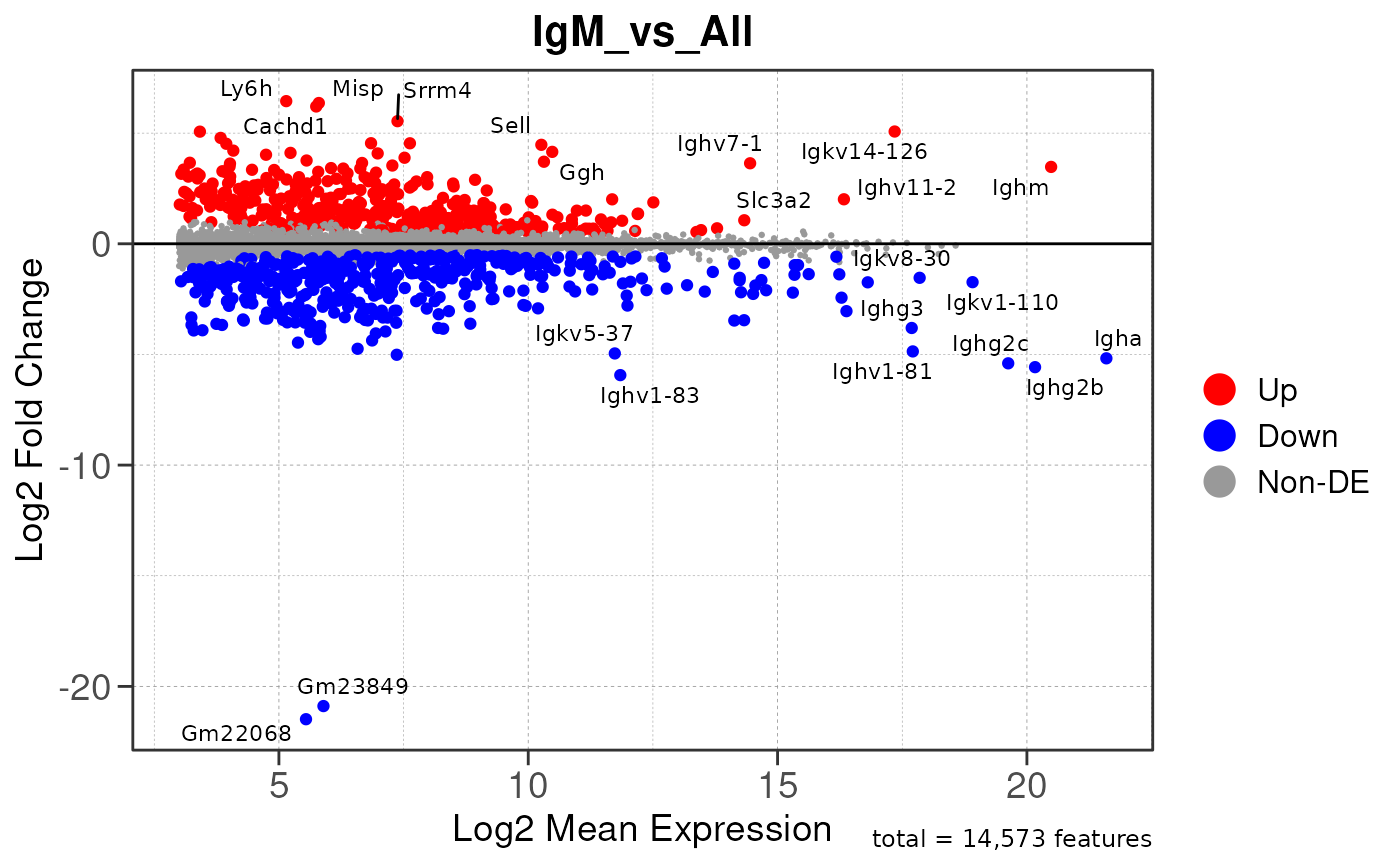
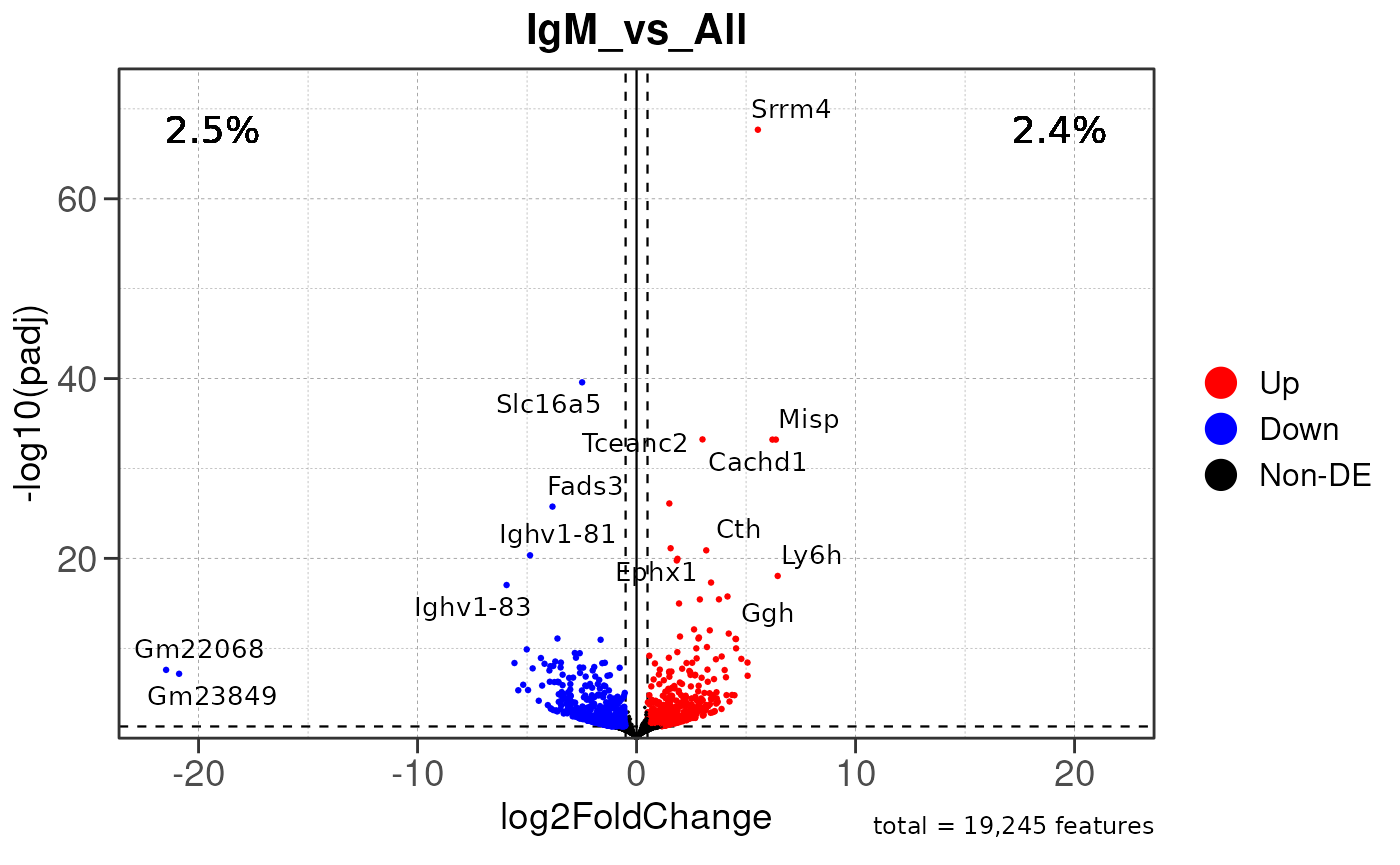
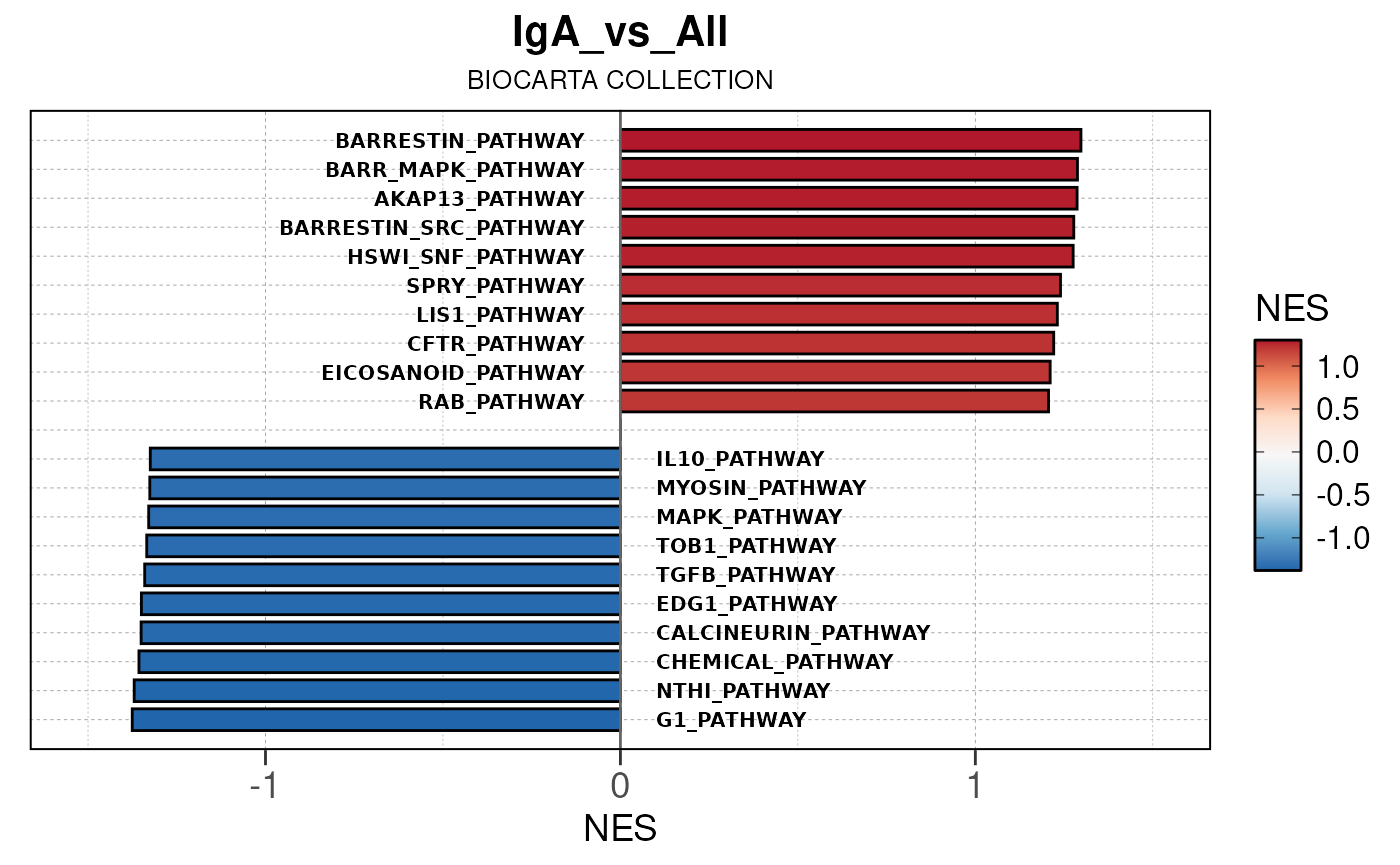
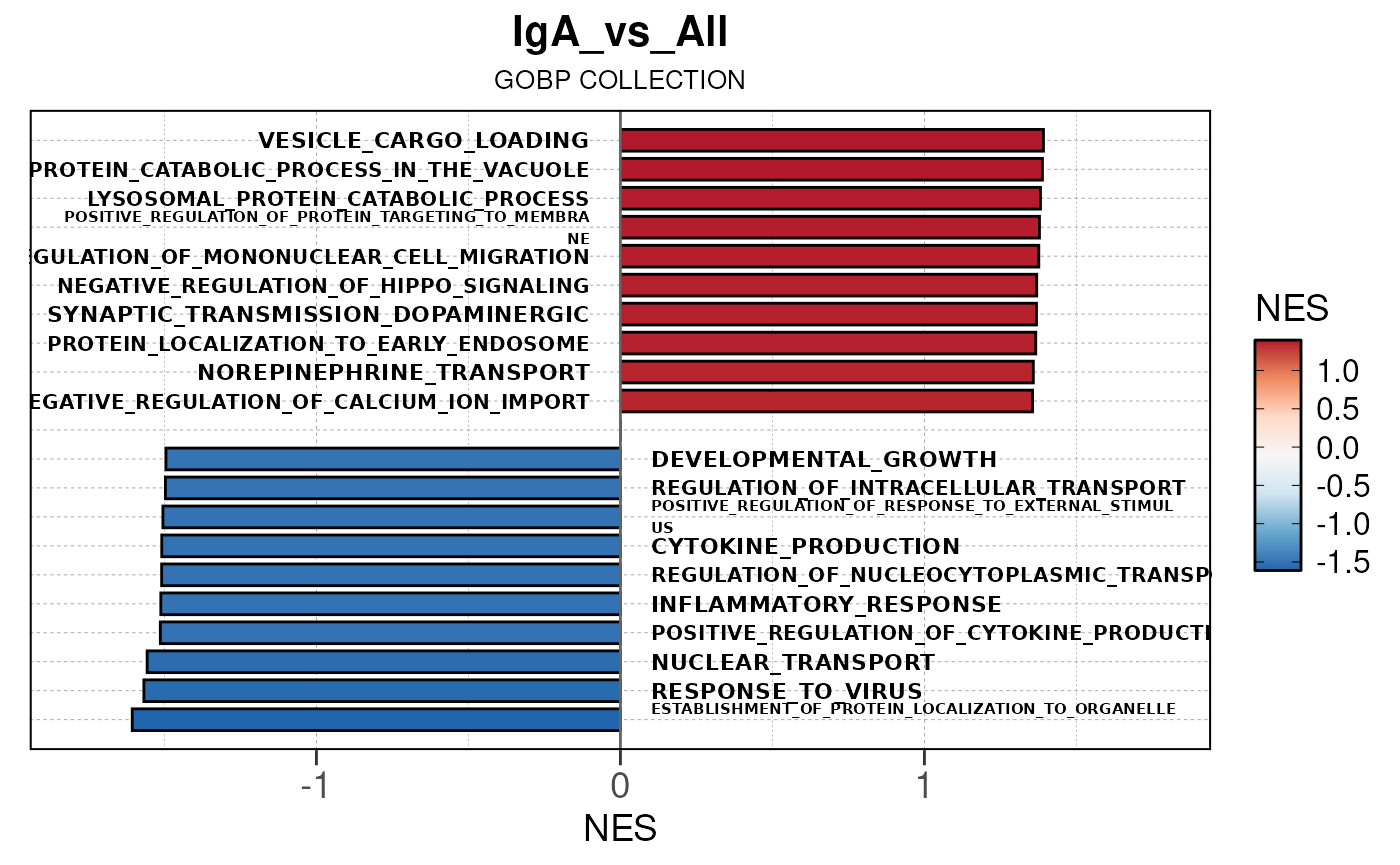
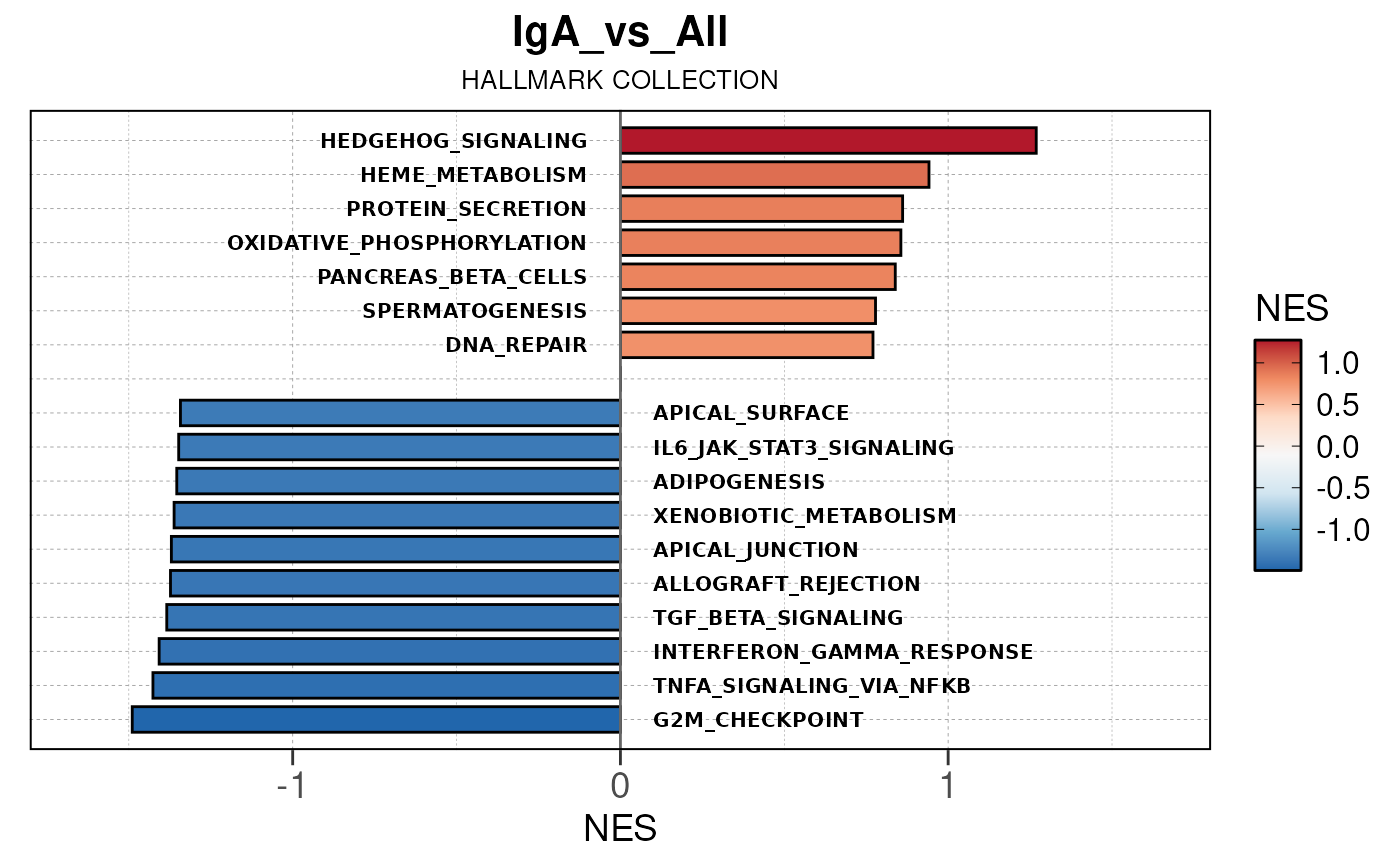
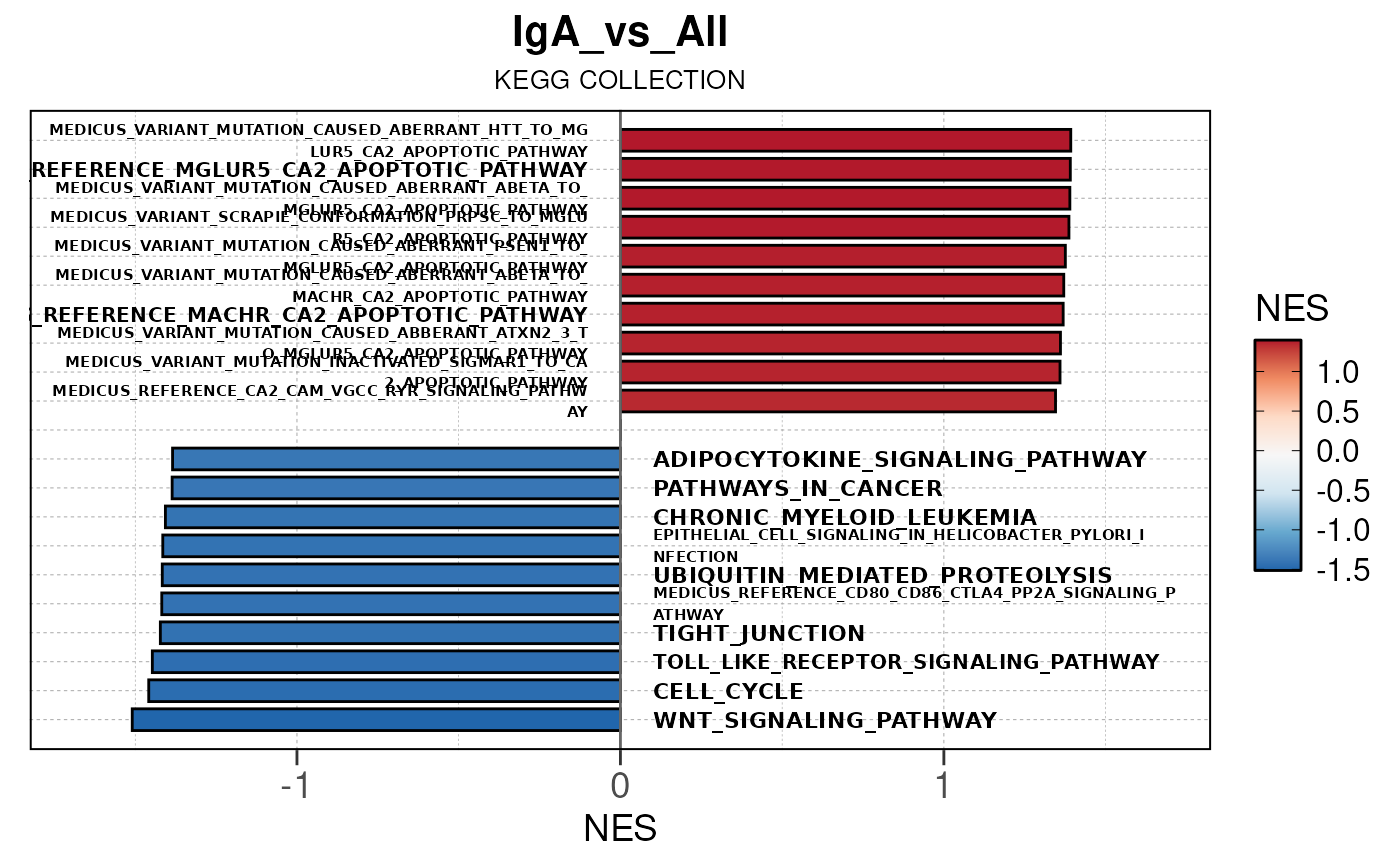
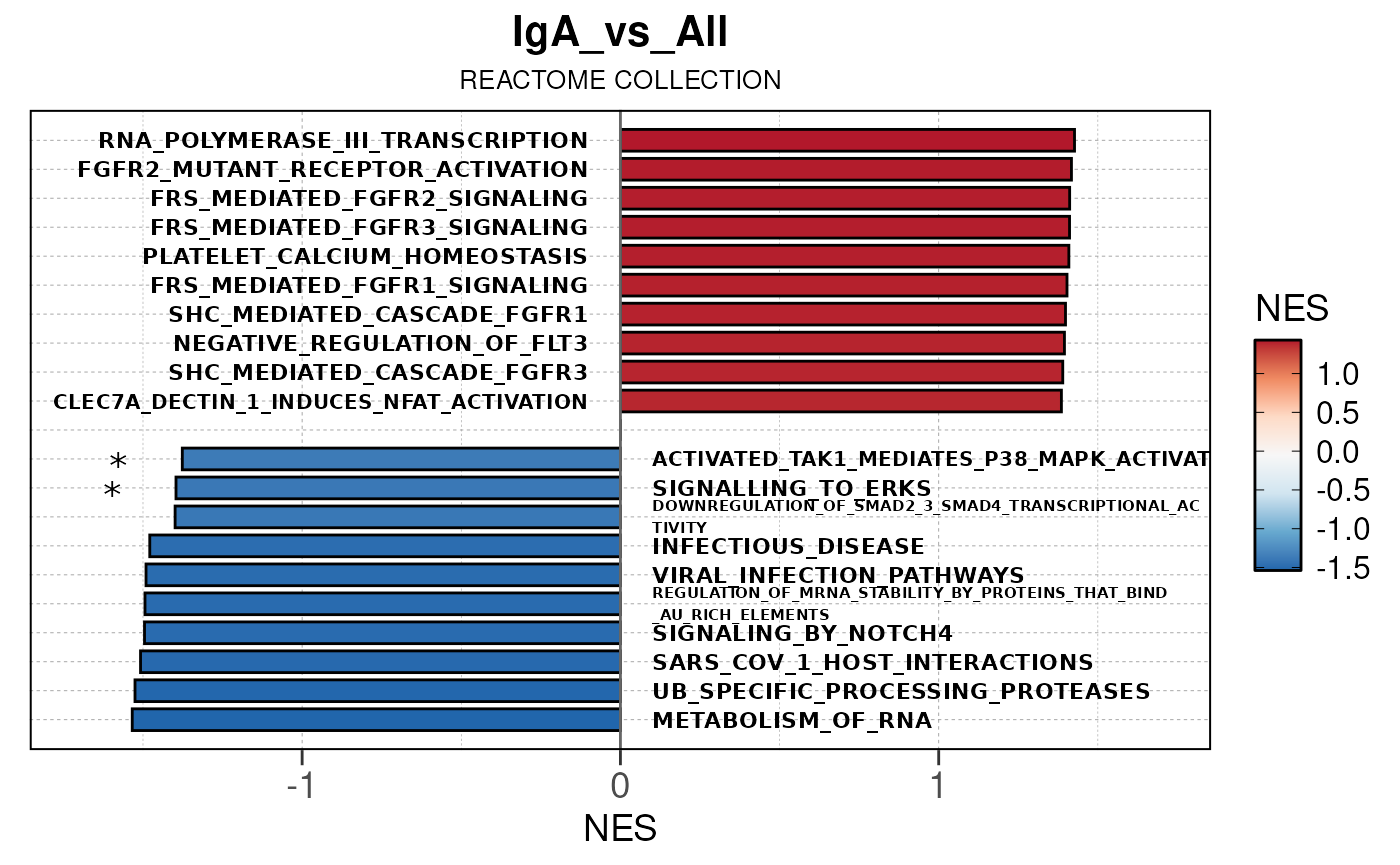
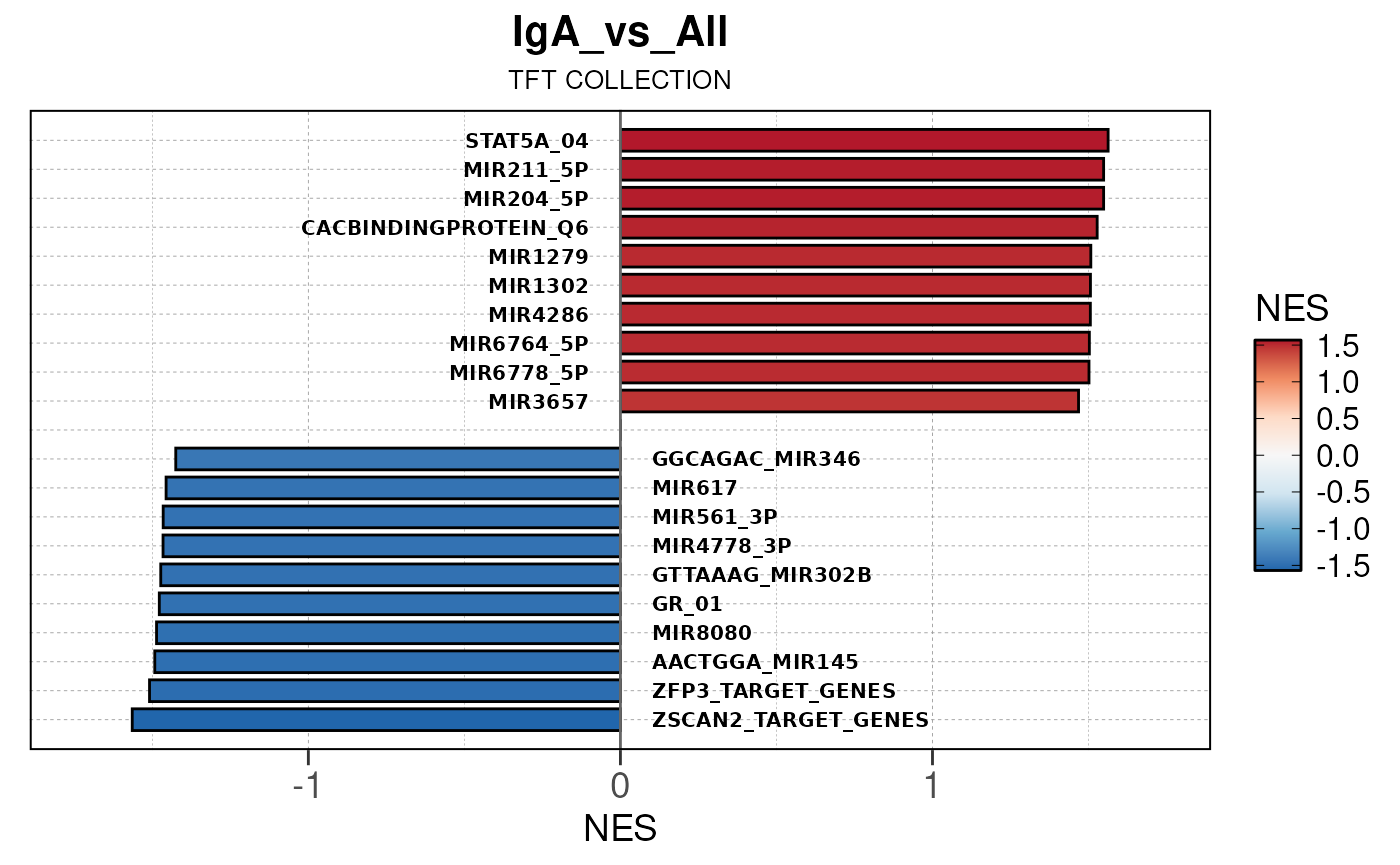
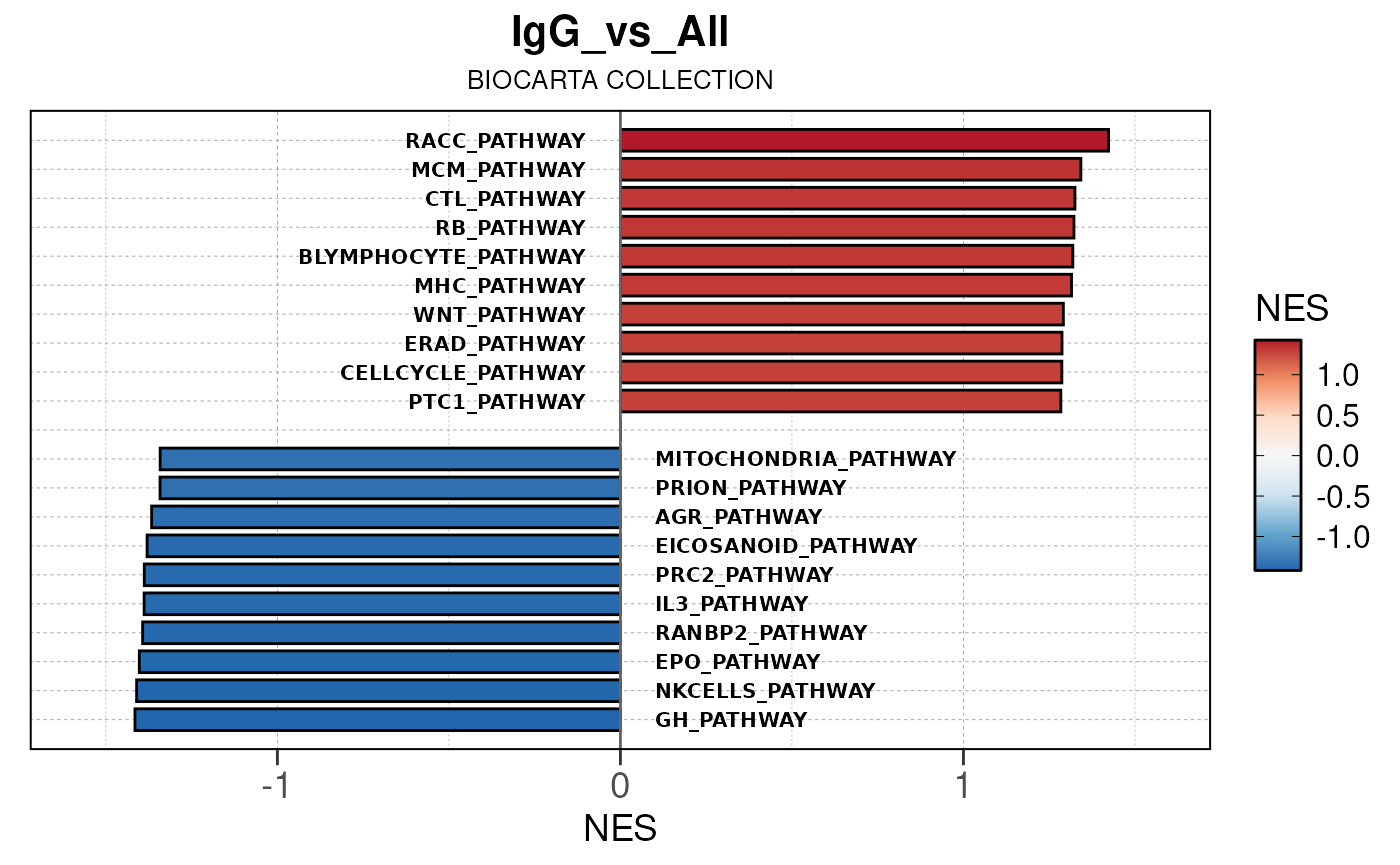
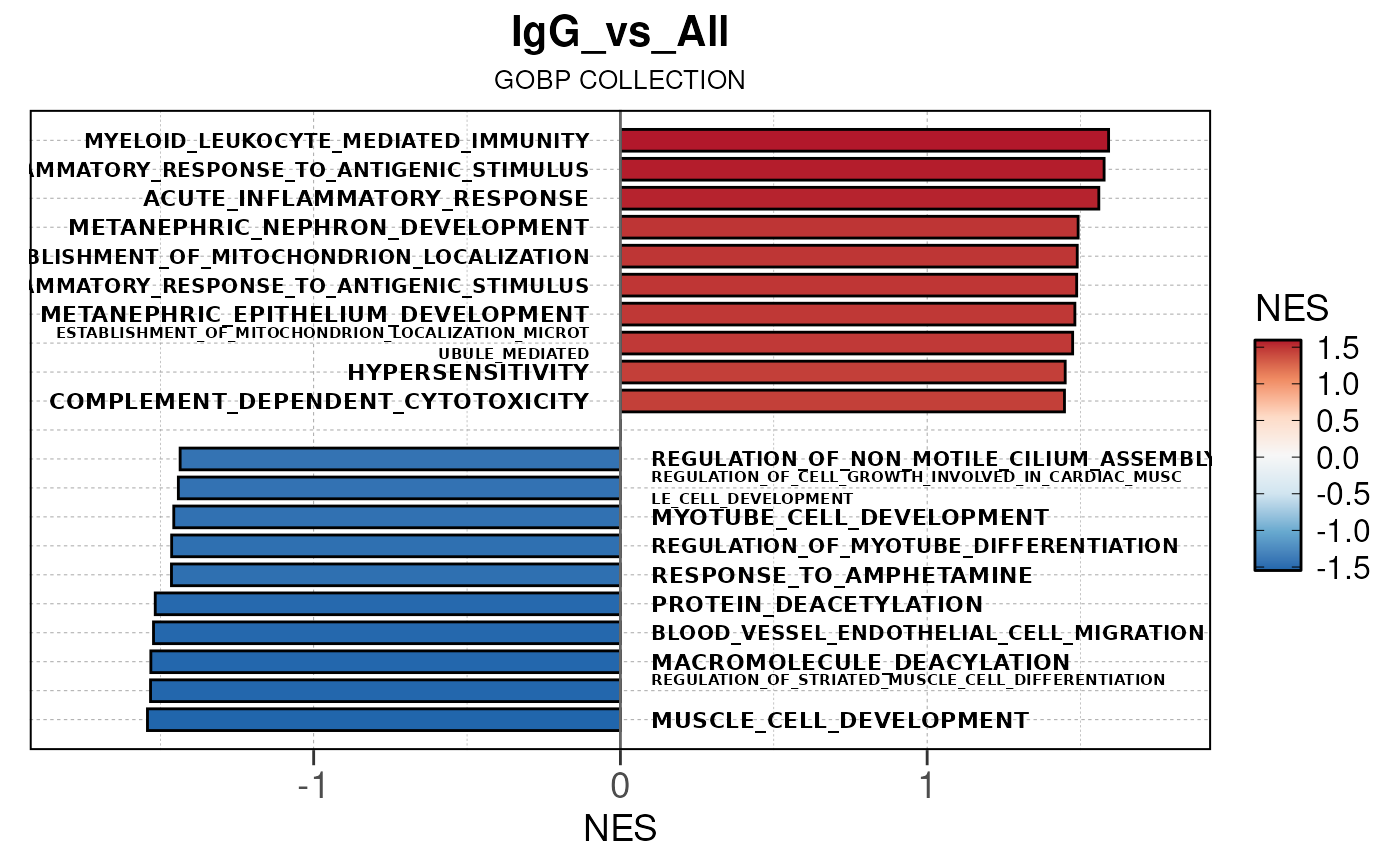
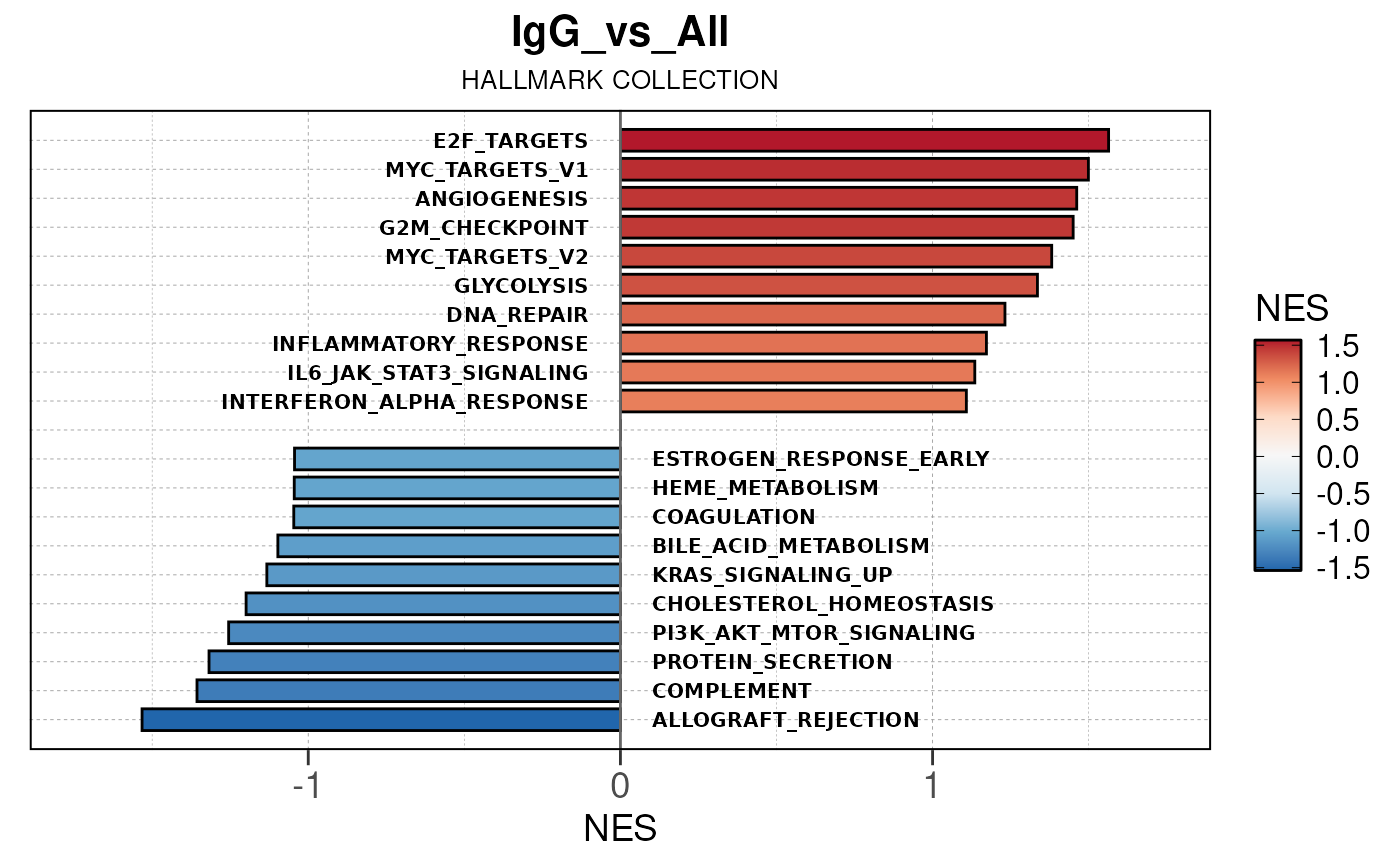
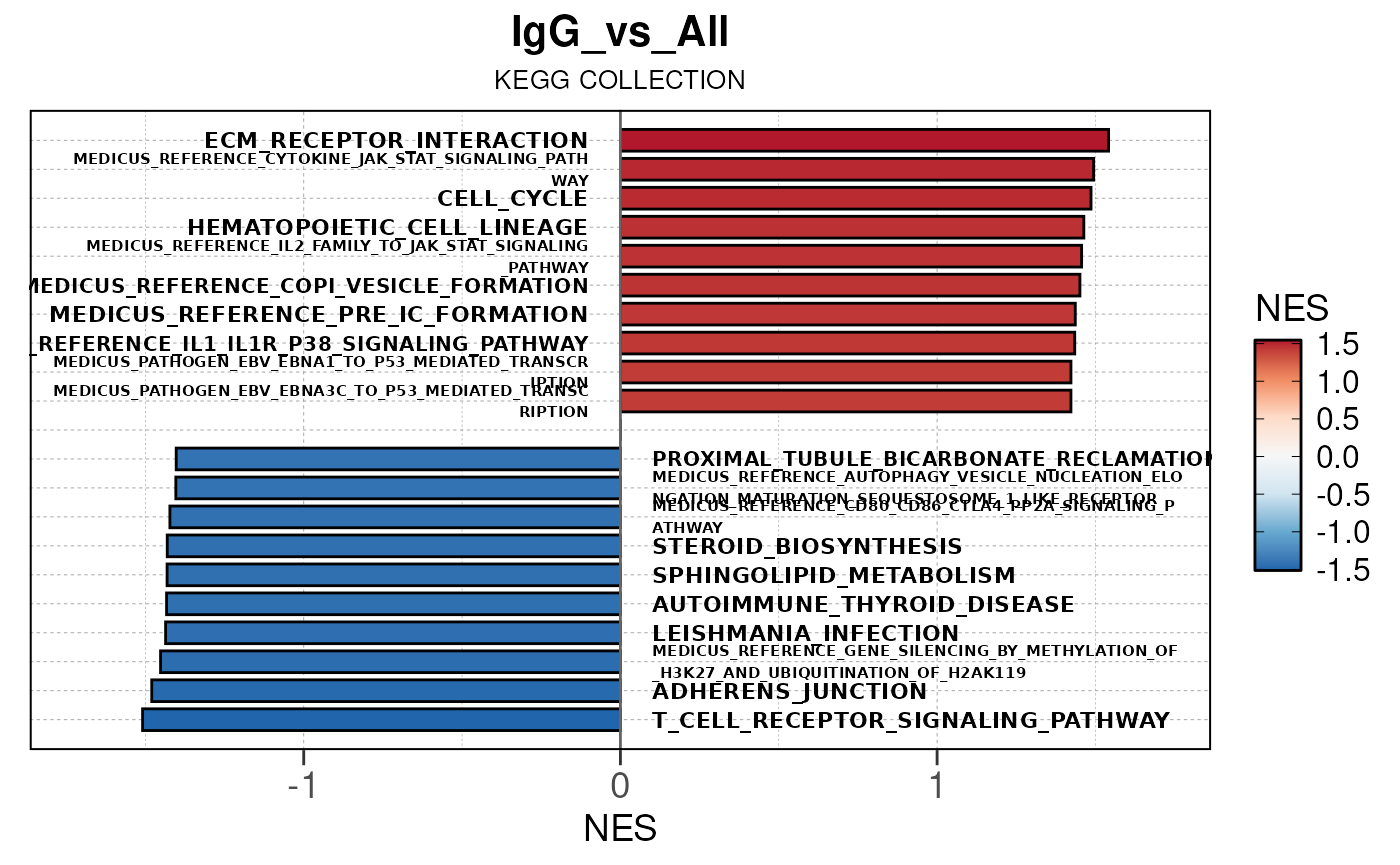
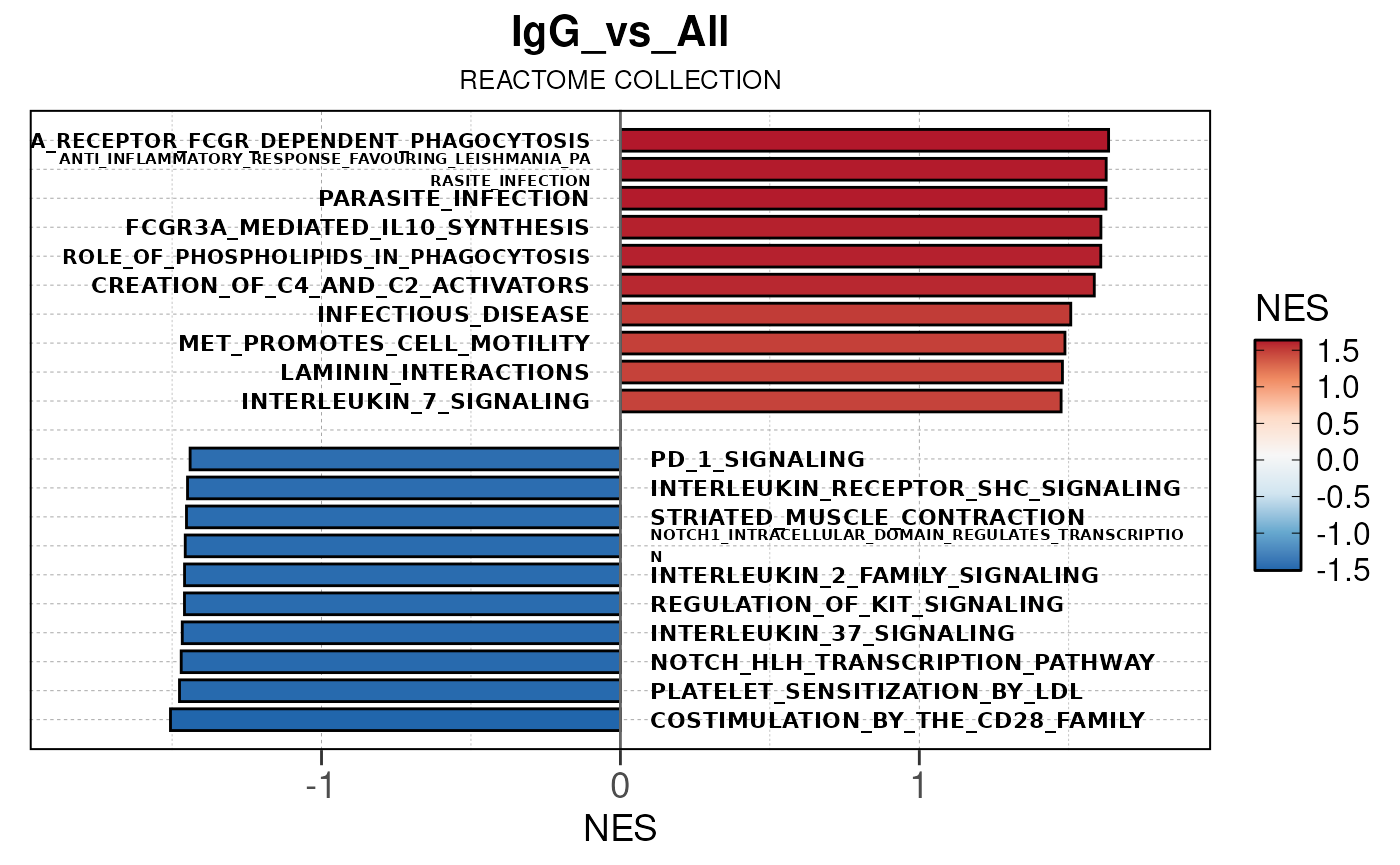
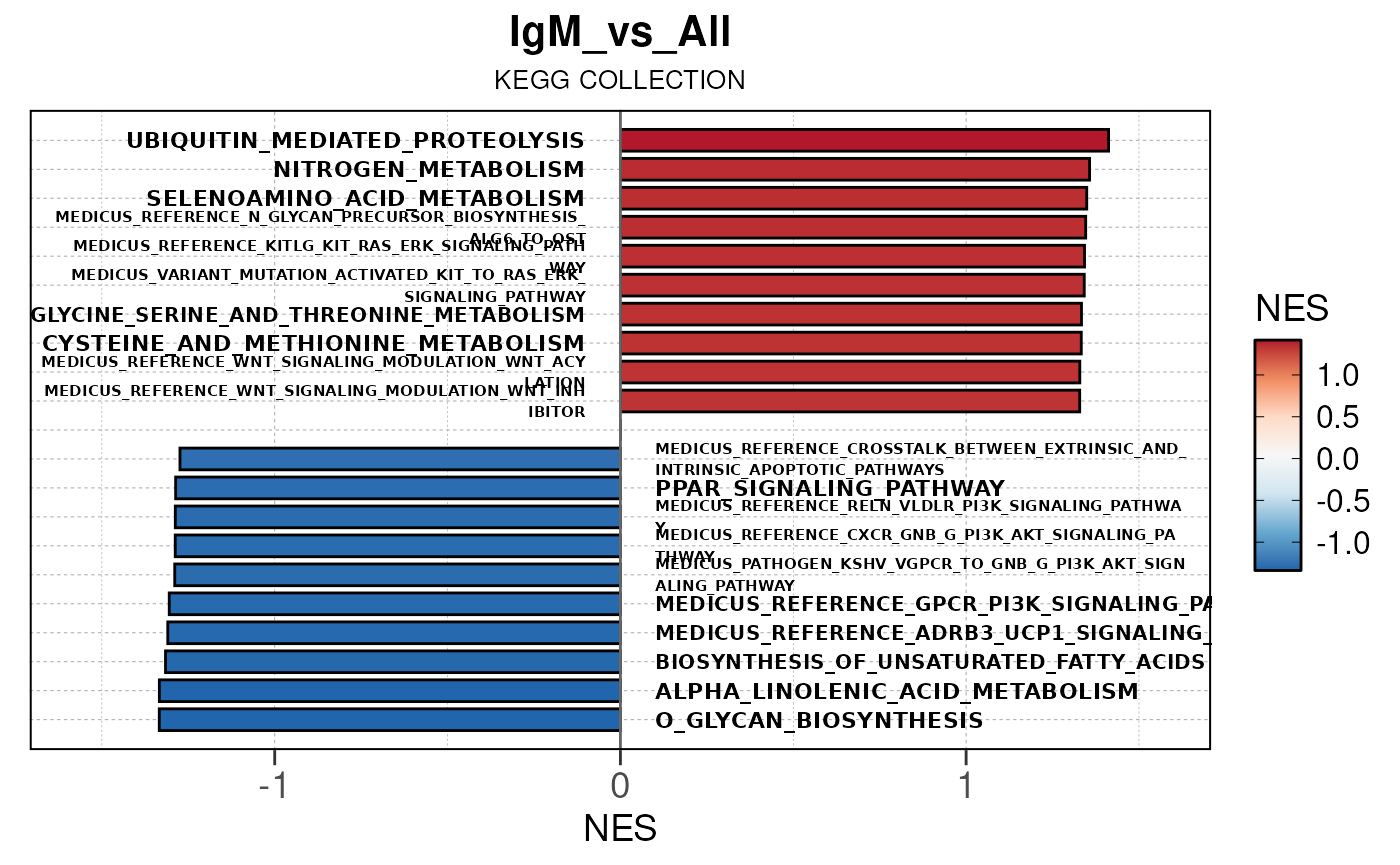
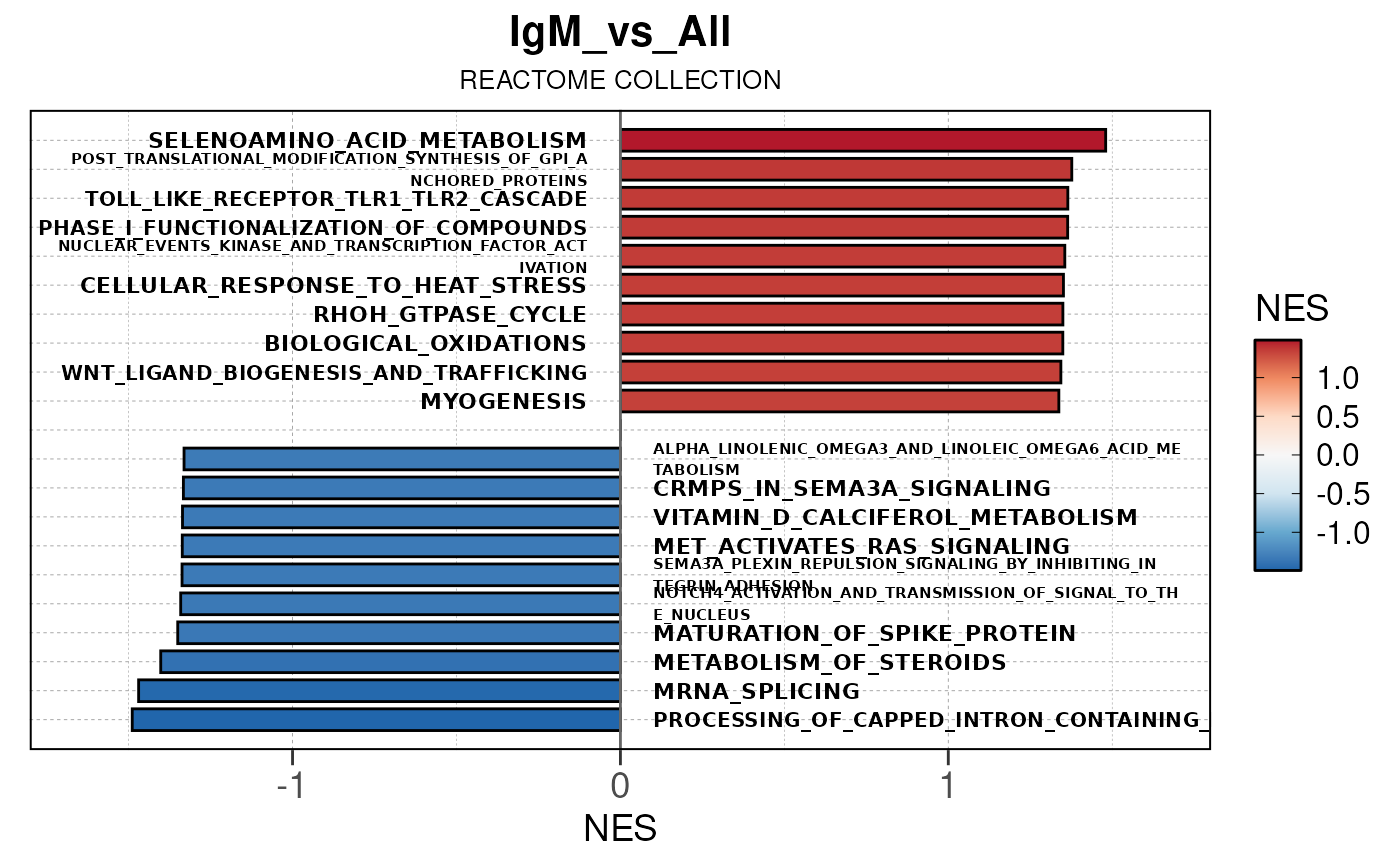
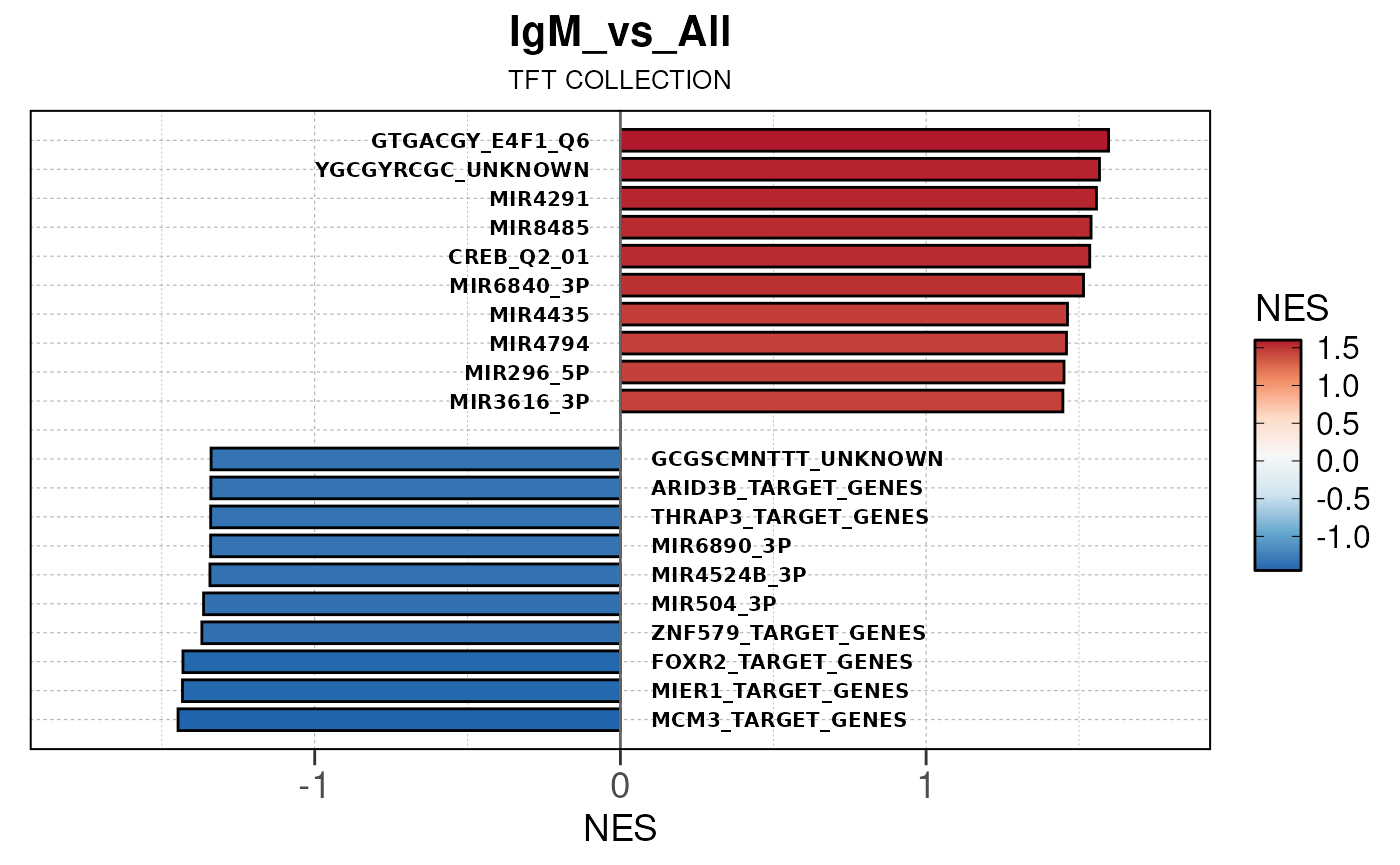
Run ATAC pipeline
# run atac pipeline
run_atac_pip(
dds,
org = "mouse",
group_by = "Group1",
remove_xy = TRUE,
remove_mt = TRUE,
quantile = 0.05,
pals = NULL,
batch = NULL,
order = "pxfc",
TSS = TRUE,
save_dir = save_dir)
## renv already initialized, restoring renv
## - The library is already synchronized with the lockfile.
## WARNING: '/tmp/RtmpZnQkvP' already exists
## group_by is not a factor, converting to factor
## group_by is not in the design, adding to design
## Running ATAC-seq pipeline with DESeq2pip v1.0.2
## Running quality control pipeline...
## Removing 3331 XY genes out of 74851 total genes...
## Removing 2 MT genes out of 71520 total genes...
## Filtering genes with low expressions...
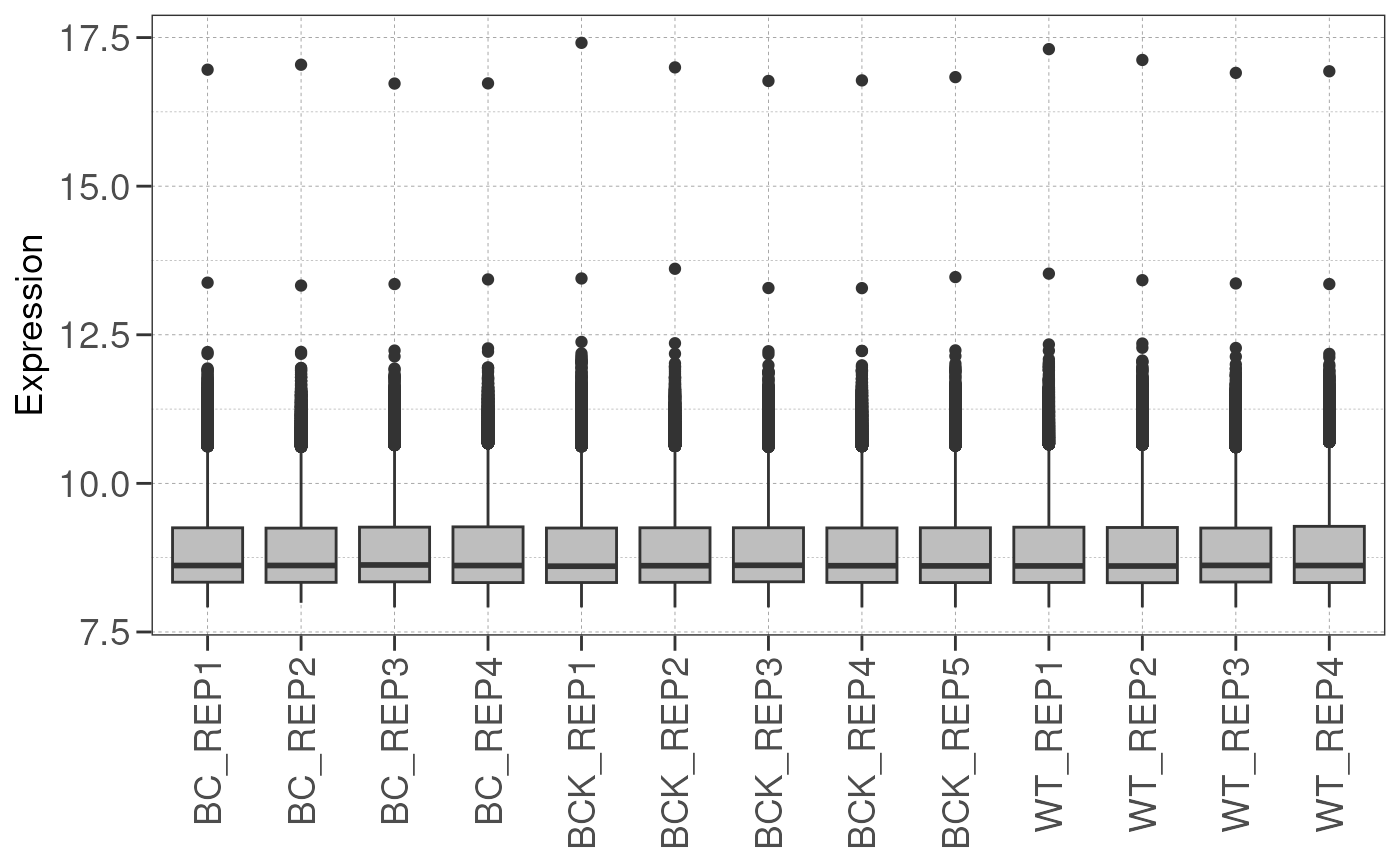
## Saving DESeq2 object & expressions...
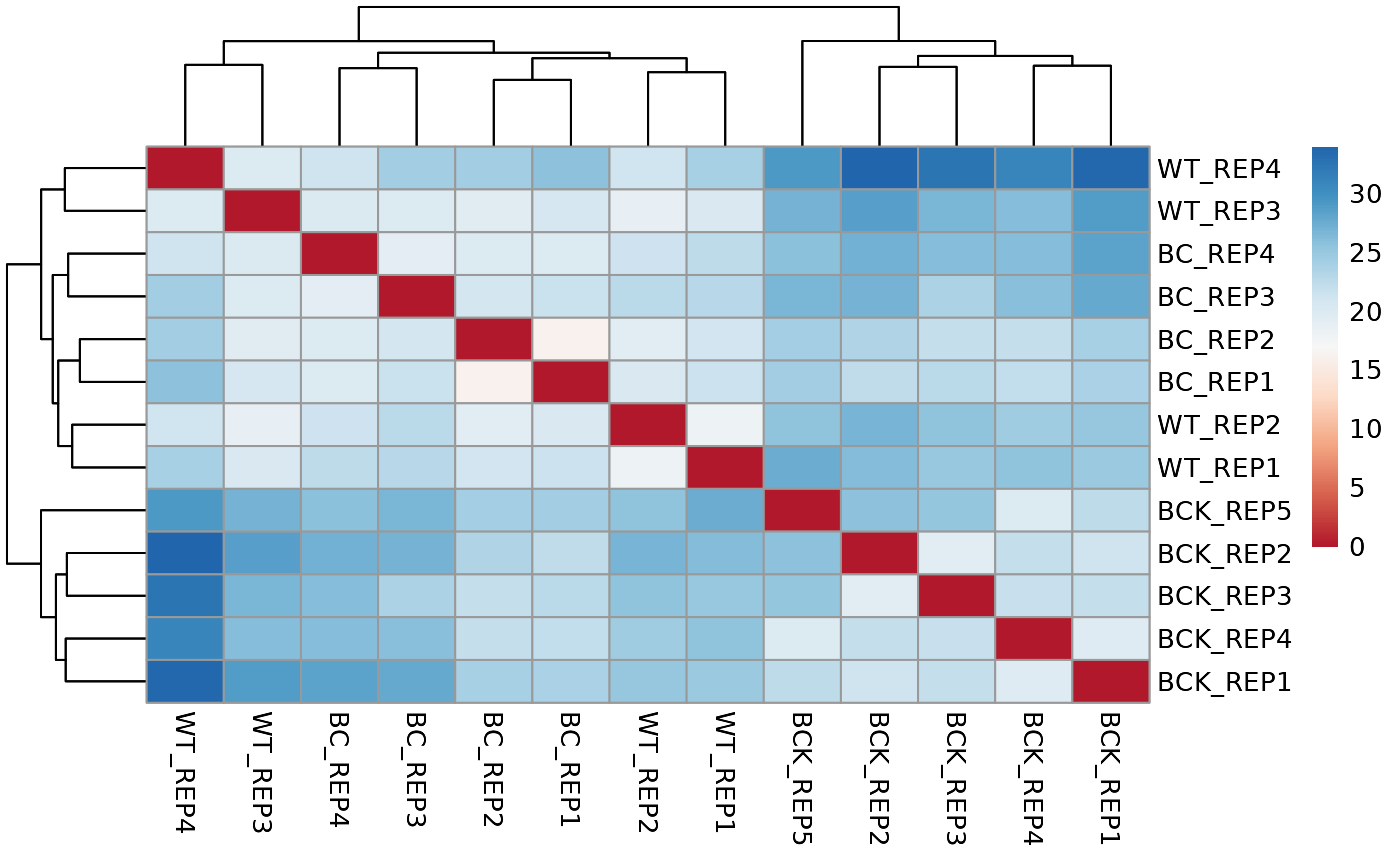
## Running distance analysis pipeline...
## Running batch correction...
## Running PCA and distance analysis...
## using ntop=500 top features by variance
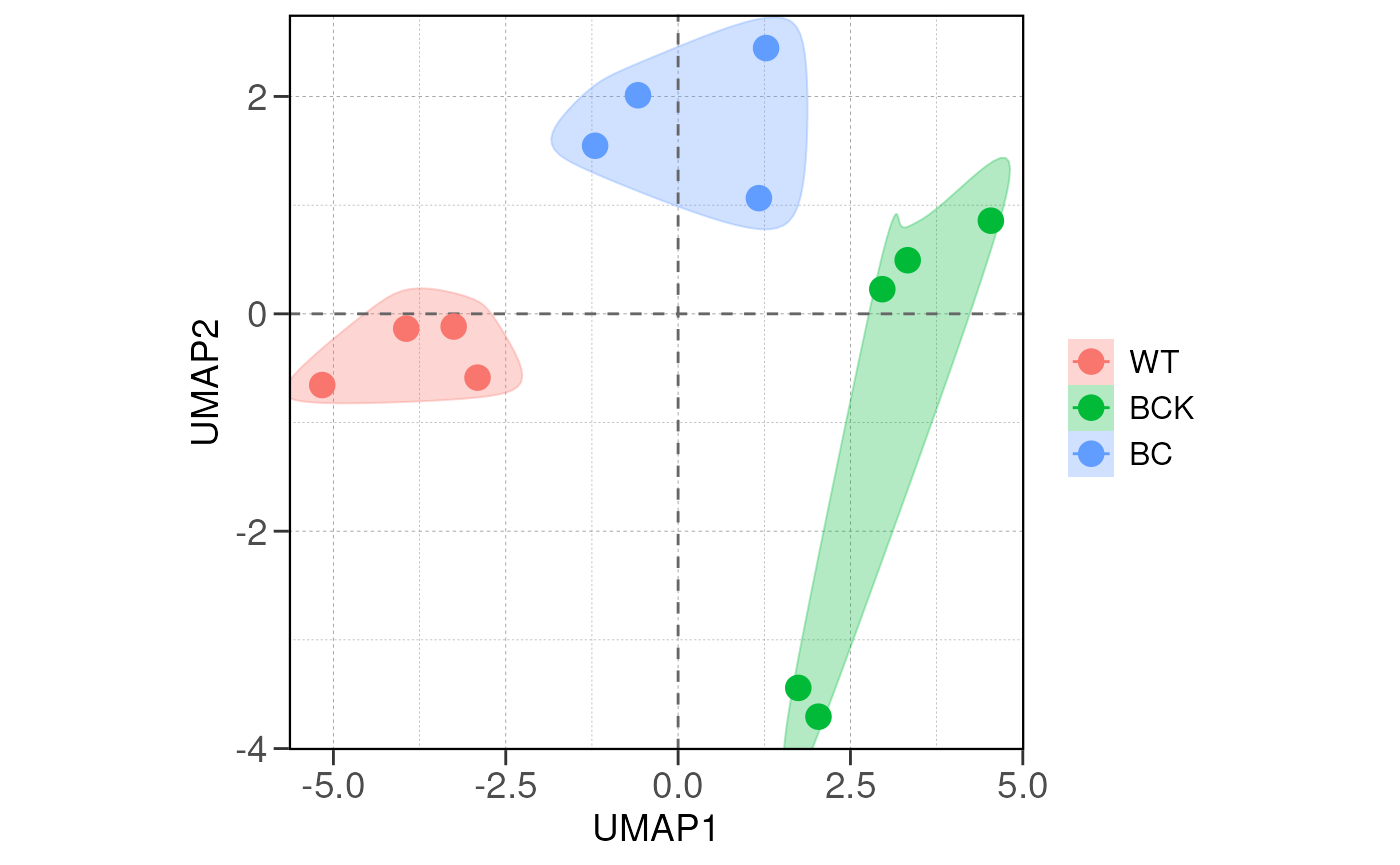
## group_by is not a factor, converting to factor
## using ntop=500 top features by variance
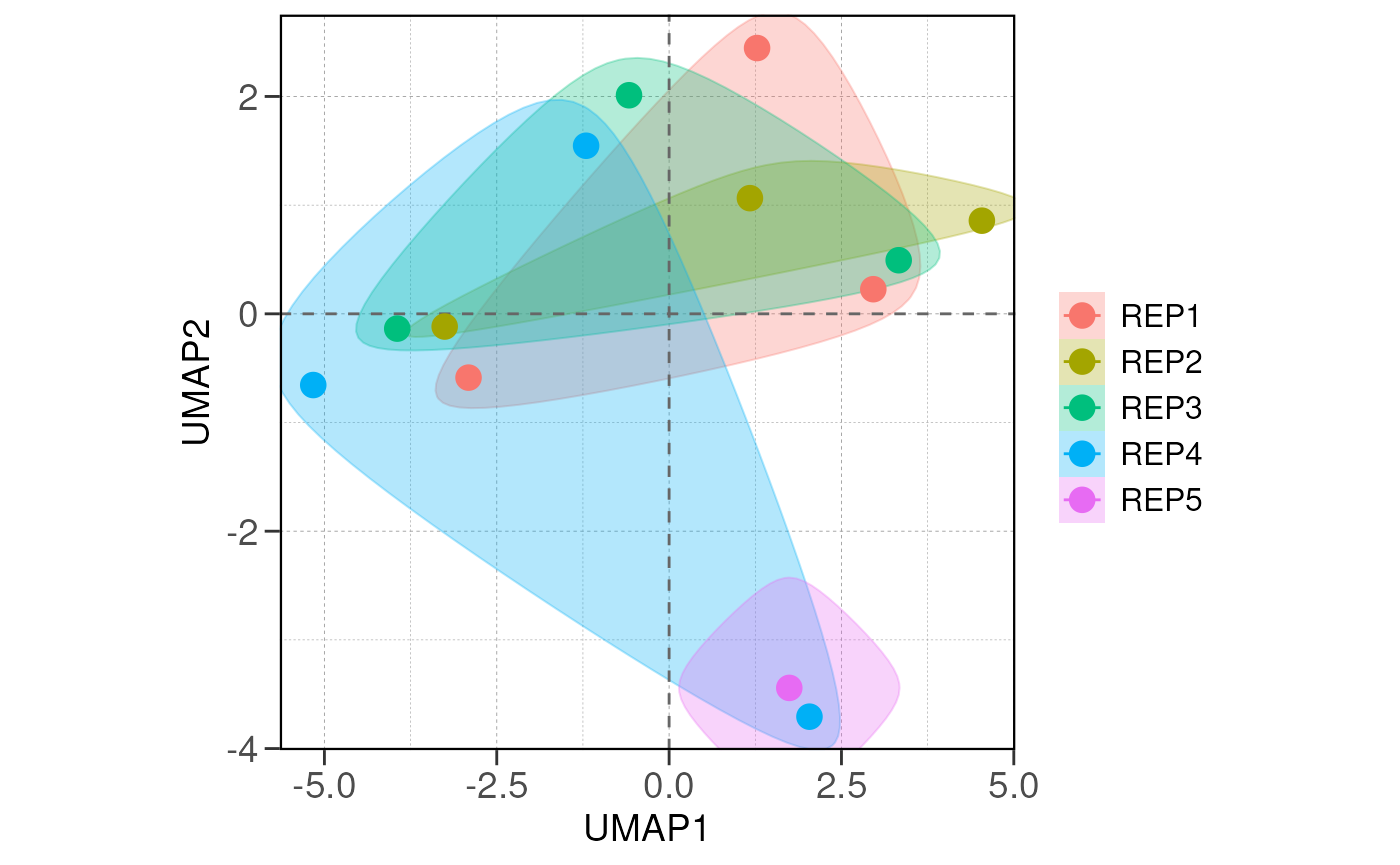
## using ntop=500 top features by variance
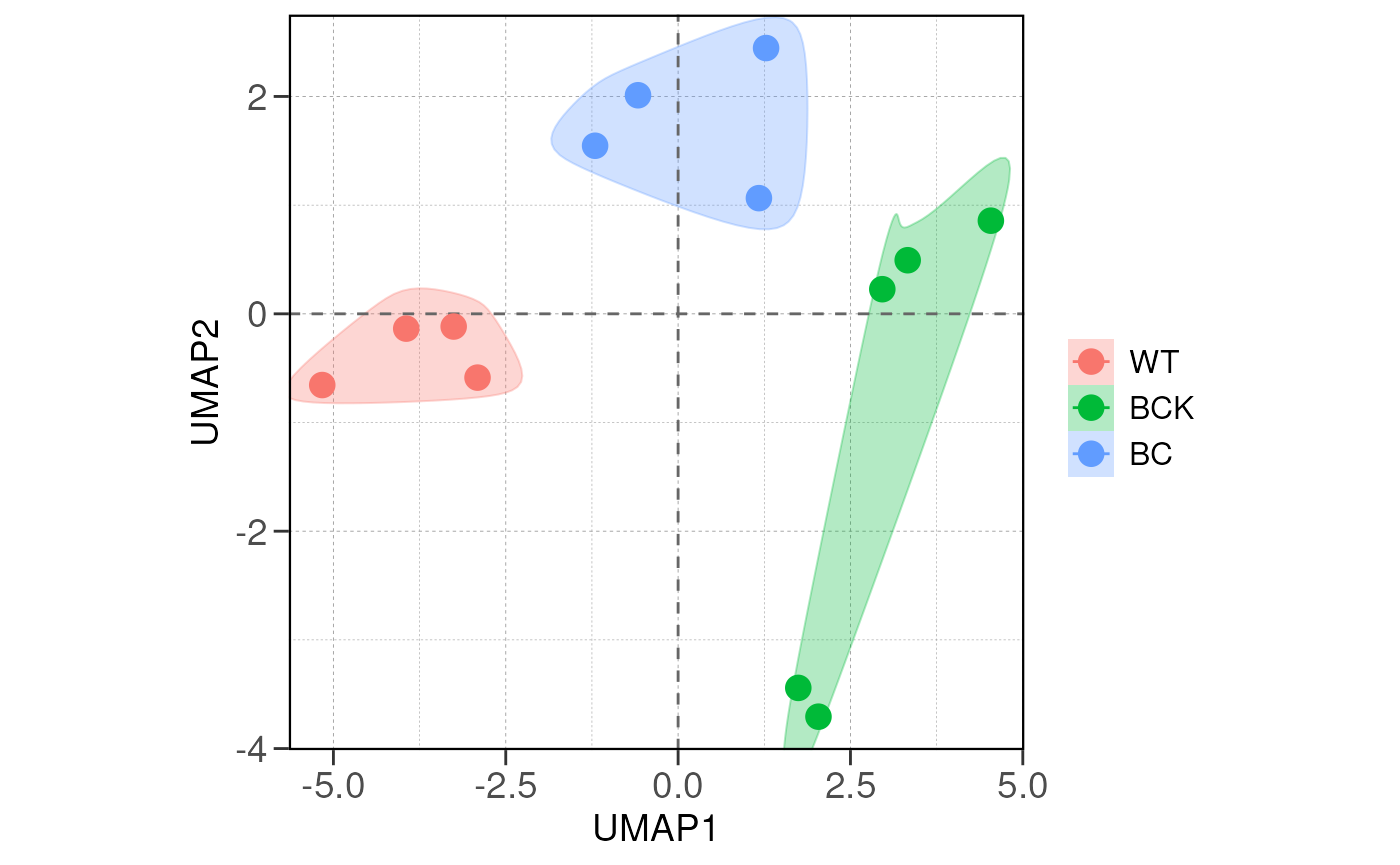
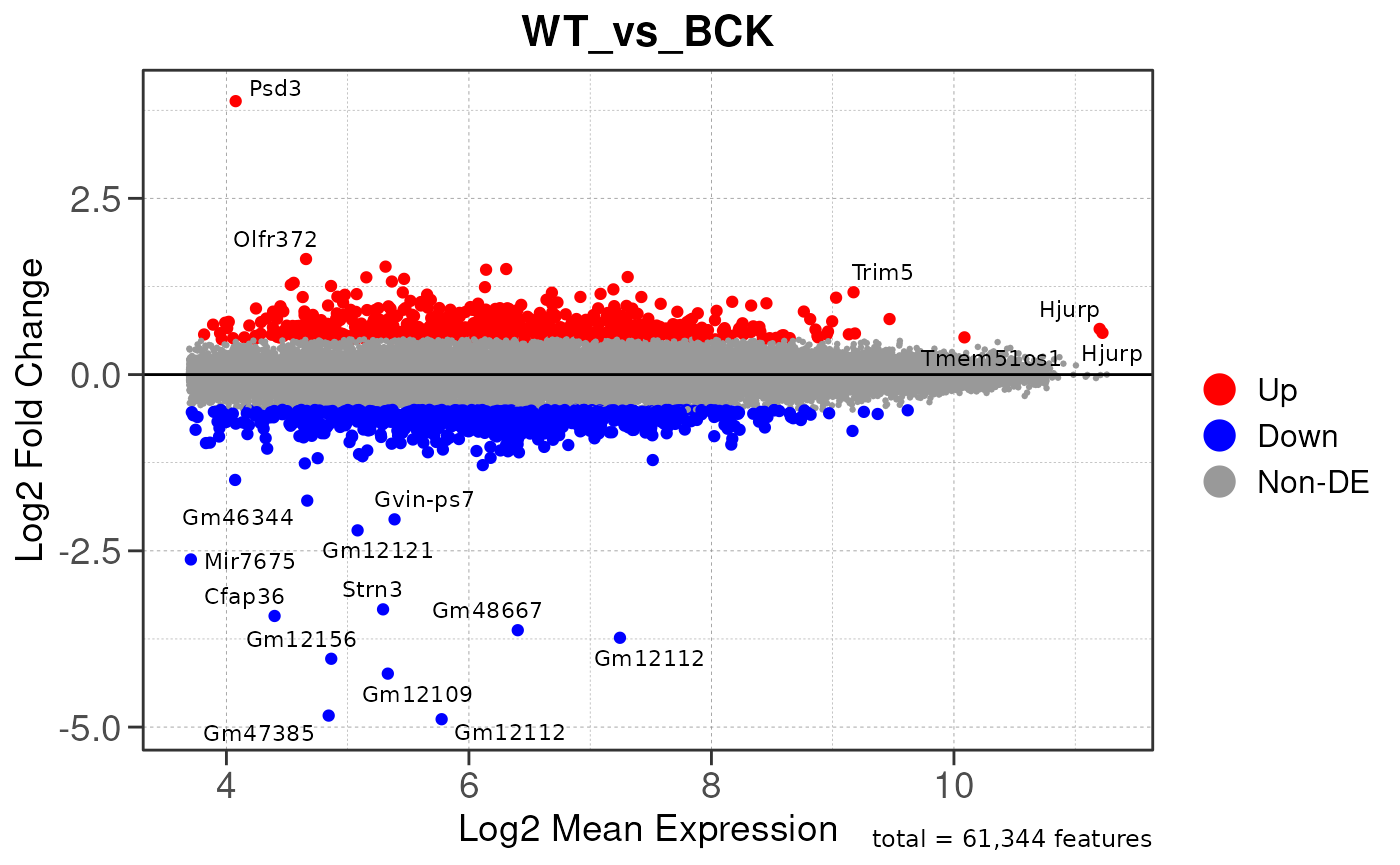
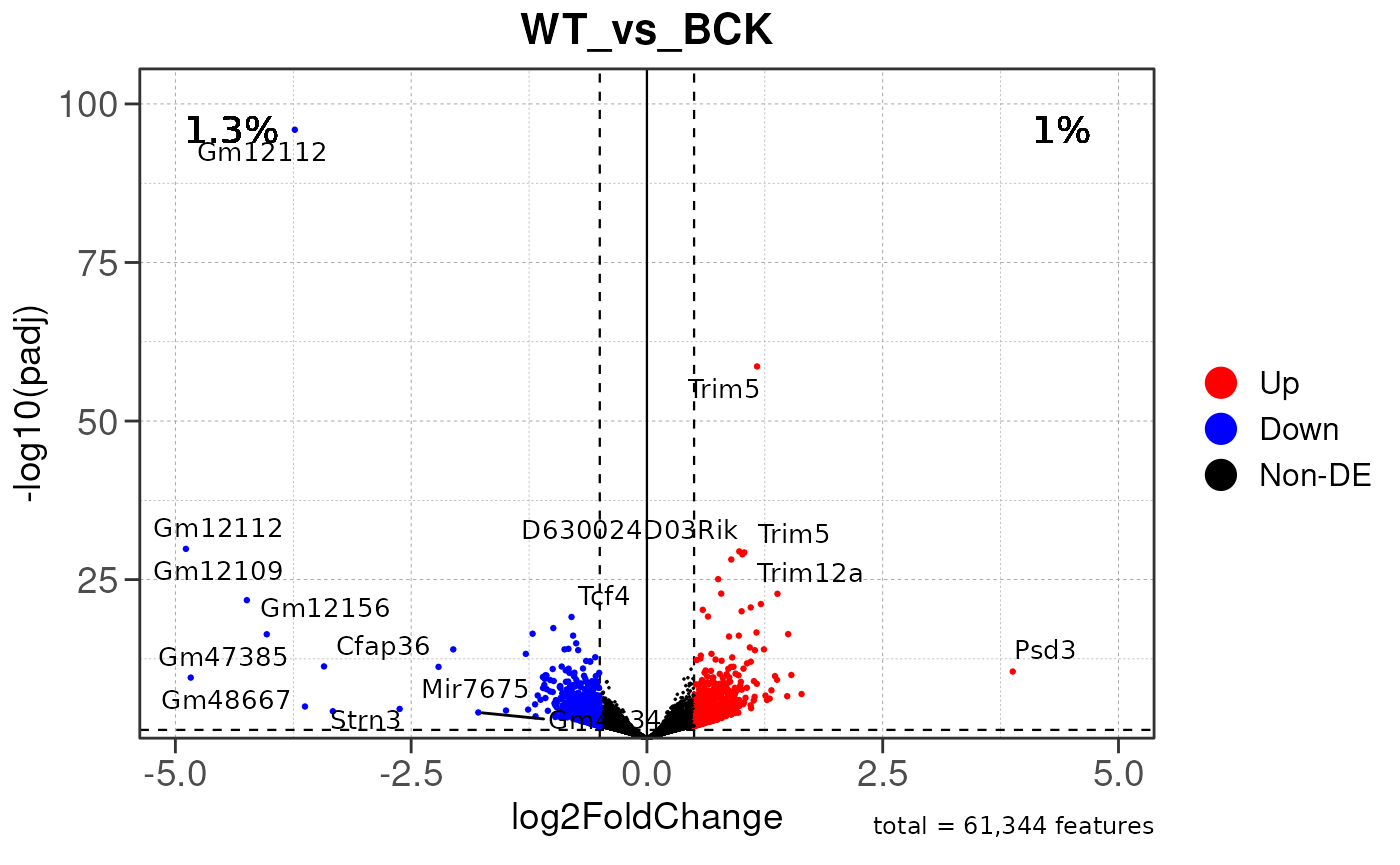
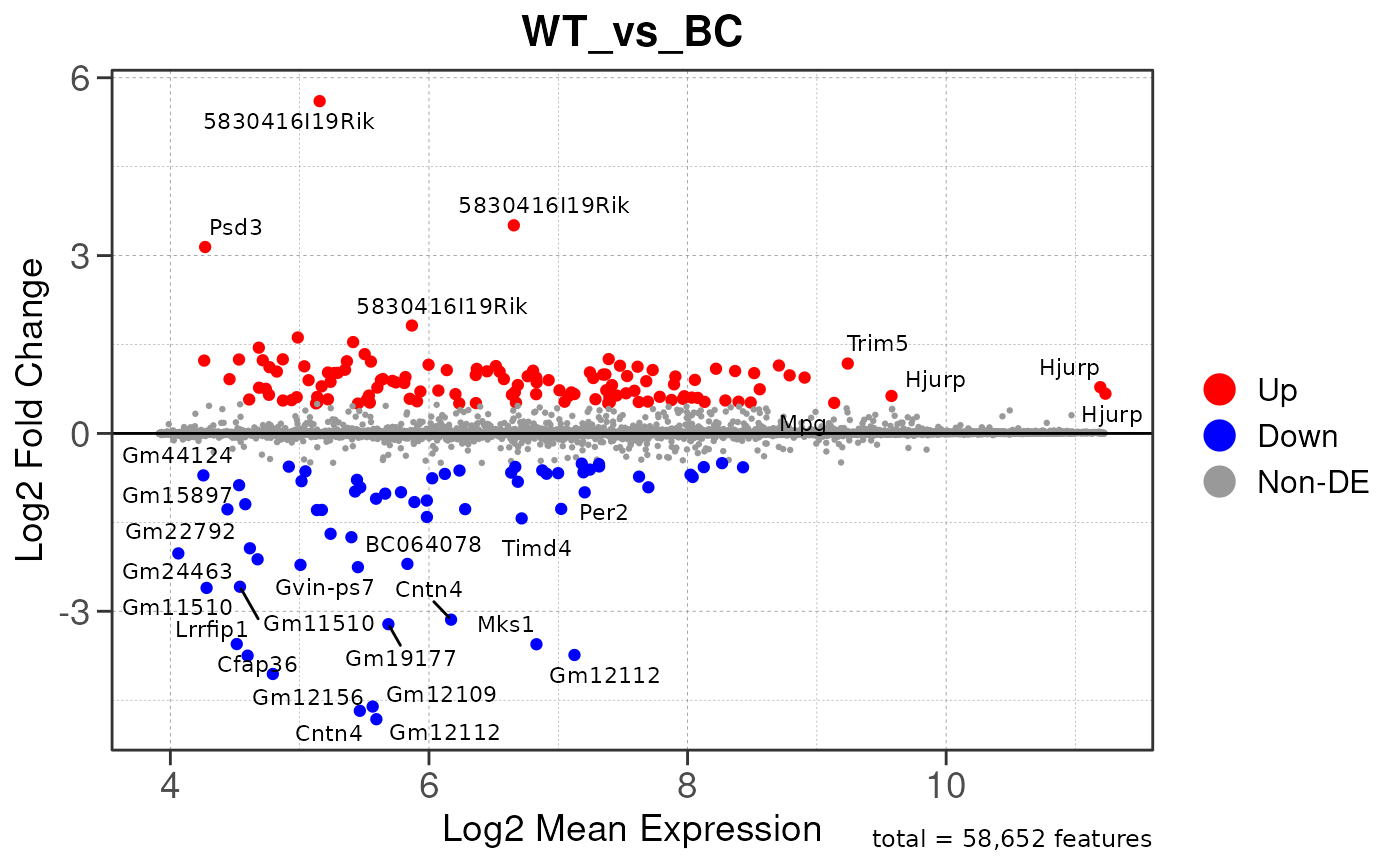
## Running PAIRWISE differential expression analysis for ALL PEAKS...
## Running PAIRWISE differential expression analysis...
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 1373 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 1373 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 59953 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 28 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 59953 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 34 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 157 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 158 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 58465 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 22 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 58465 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 23 unlabeled data points (too many overlaps). Consider increasing max.overlaps
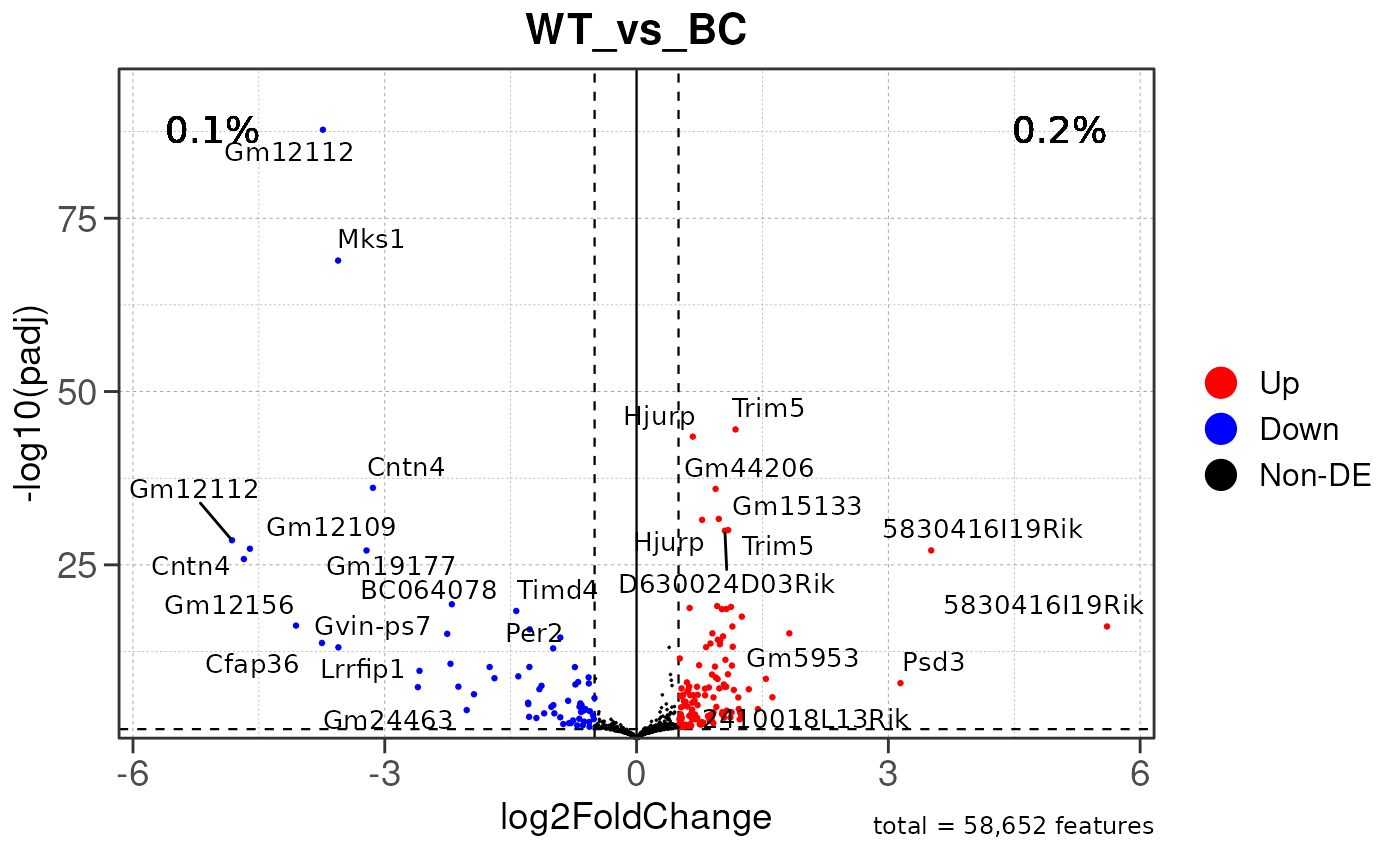
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 180 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 180 unlabeled data points (too many overlaps). Consider increasing max.overlaps
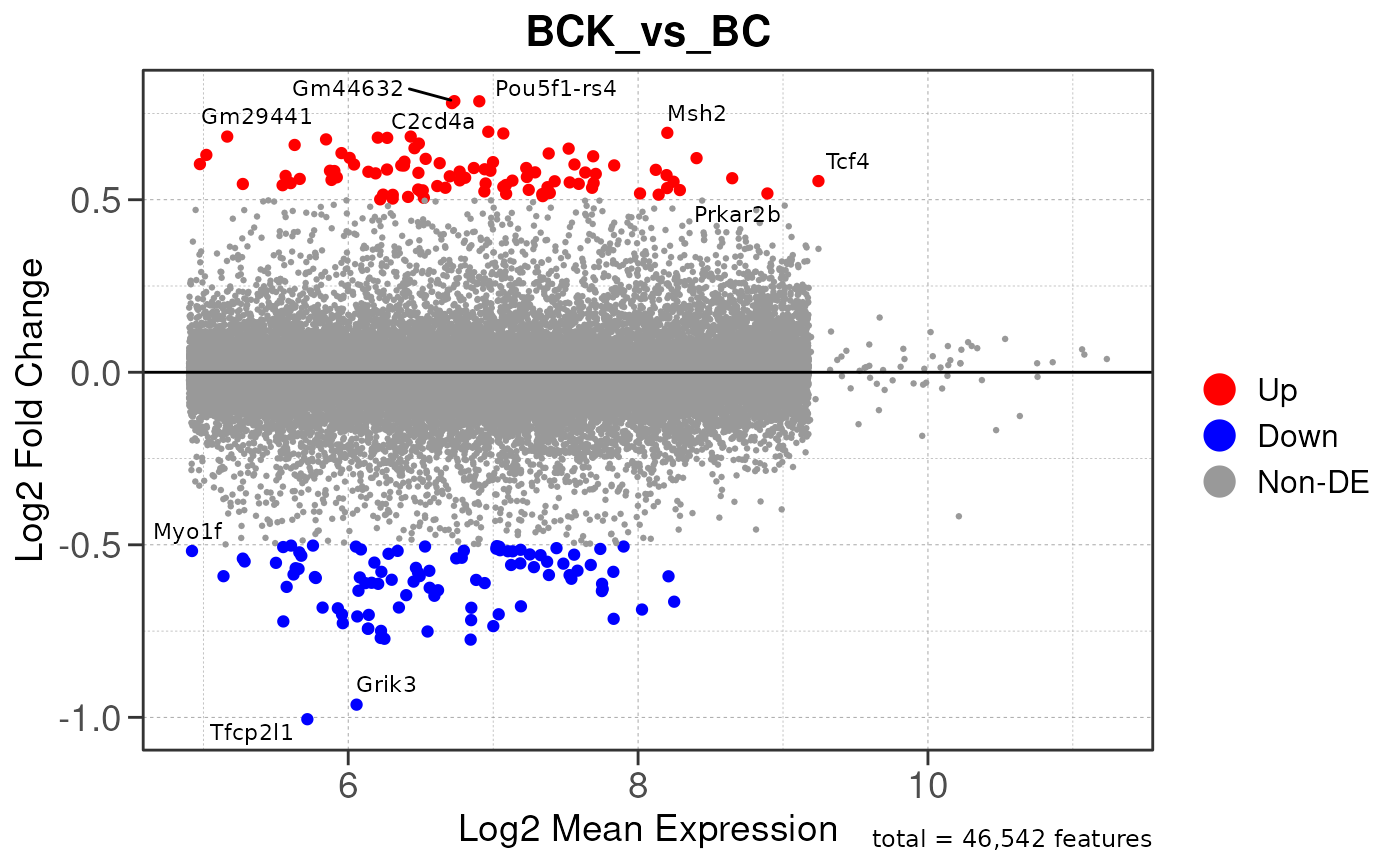
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 46352 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 17 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 46352 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 33 unlabeled data points (too many overlaps). Consider increasing max.overlaps
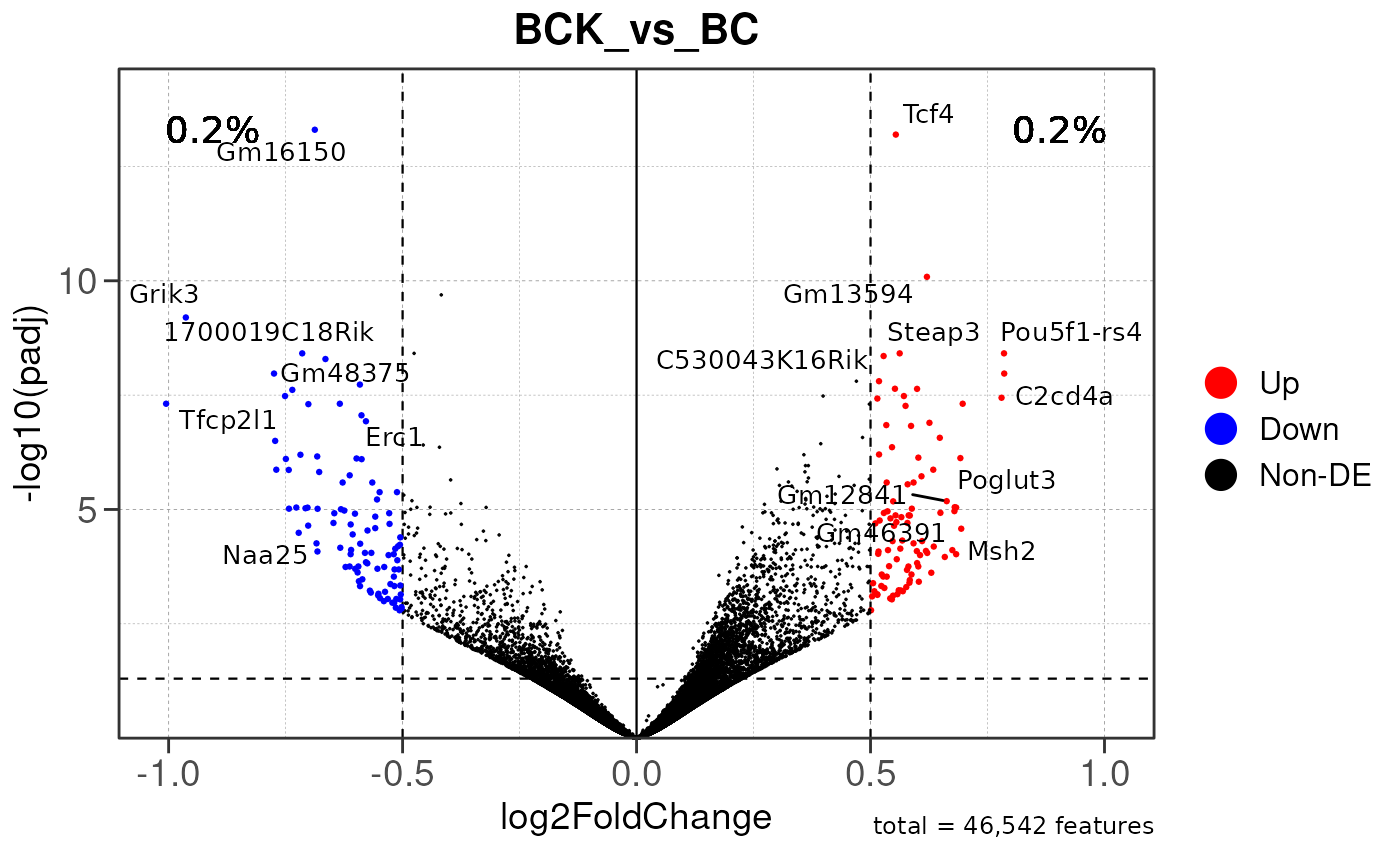
## Generating PAIRWISE peak annotation plots...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 1391 peaks in All DE Peaks...
## Creating pie charts for 623 peaks in Upregulated DE Peaks...
## Creating pie charts for 768 peaks in Downregulated DE Peaks...
## Saving pie charts to PDF...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 187 peaks in All DE Peaks...
## Creating pie charts for 124 peaks in Upregulated DE Peaks...
## Creating pie charts for 63 peaks in Downregulated DE Peaks...
## Saving pie charts to PDF...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 190 peaks in All DE Peaks...
## Creating pie charts for 93 peaks in Upregulated DE Peaks...
## Creating pie charts for 97 peaks in Downregulated DE Peaks...
## Saving pie charts to PDF...
## Running ONE-TO-ALL differential expression analysis for ALL PEAKS...
## Running ONE-TO-ALL differential expression analysis...
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## -- replacing outliers and refitting for 2 genes
## -- DESeq argument 'minReplicatesForReplace' = 7
## -- original counts are preserved in counts(dds)
## estimating dispersions
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 210 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 212 unlabeled data points (too many overlaps). Consider increasing max.overlaps
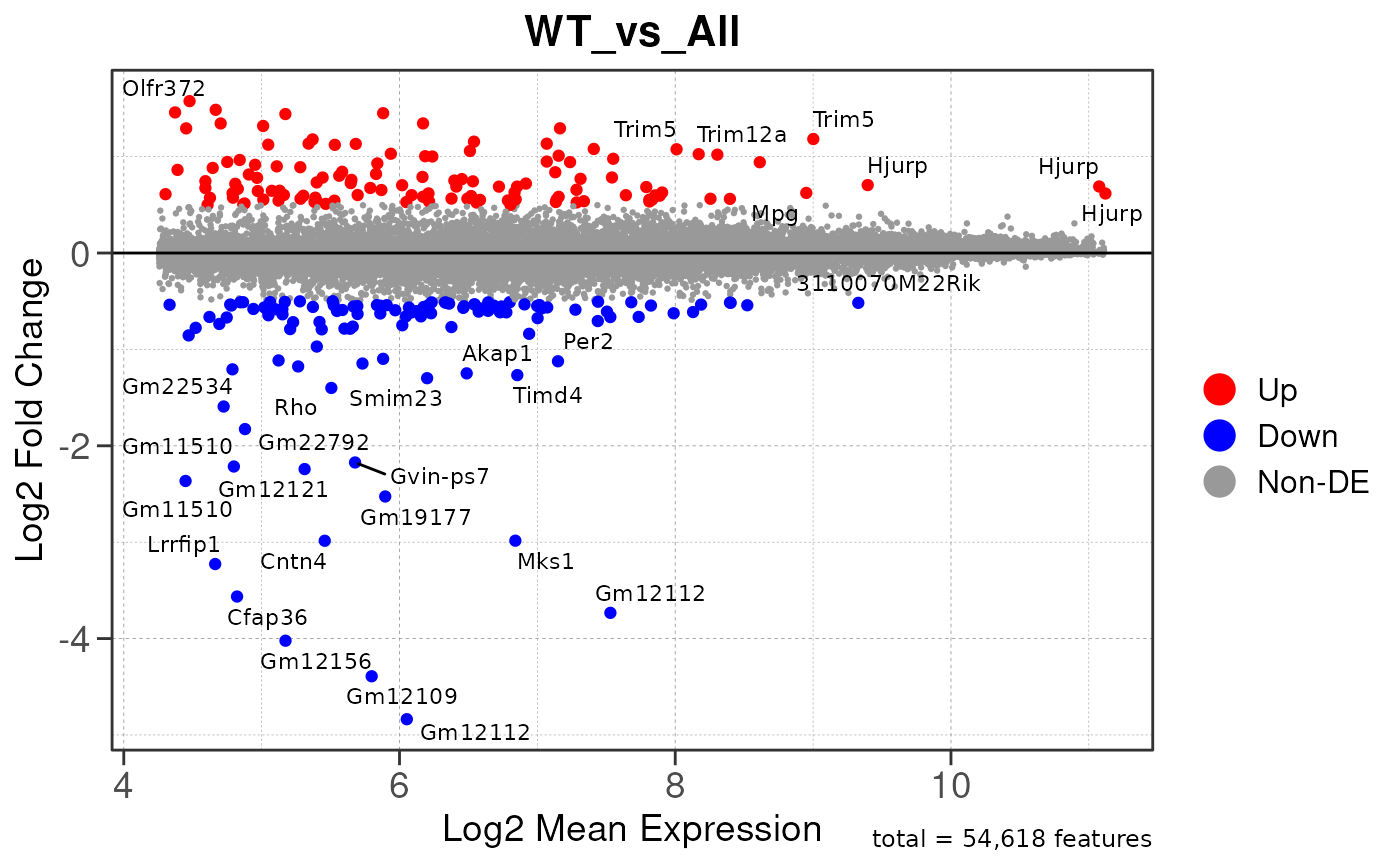
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 54377 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 29 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 54377 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 33 unlabeled data points (too many overlaps). Consider increasing max.overlaps
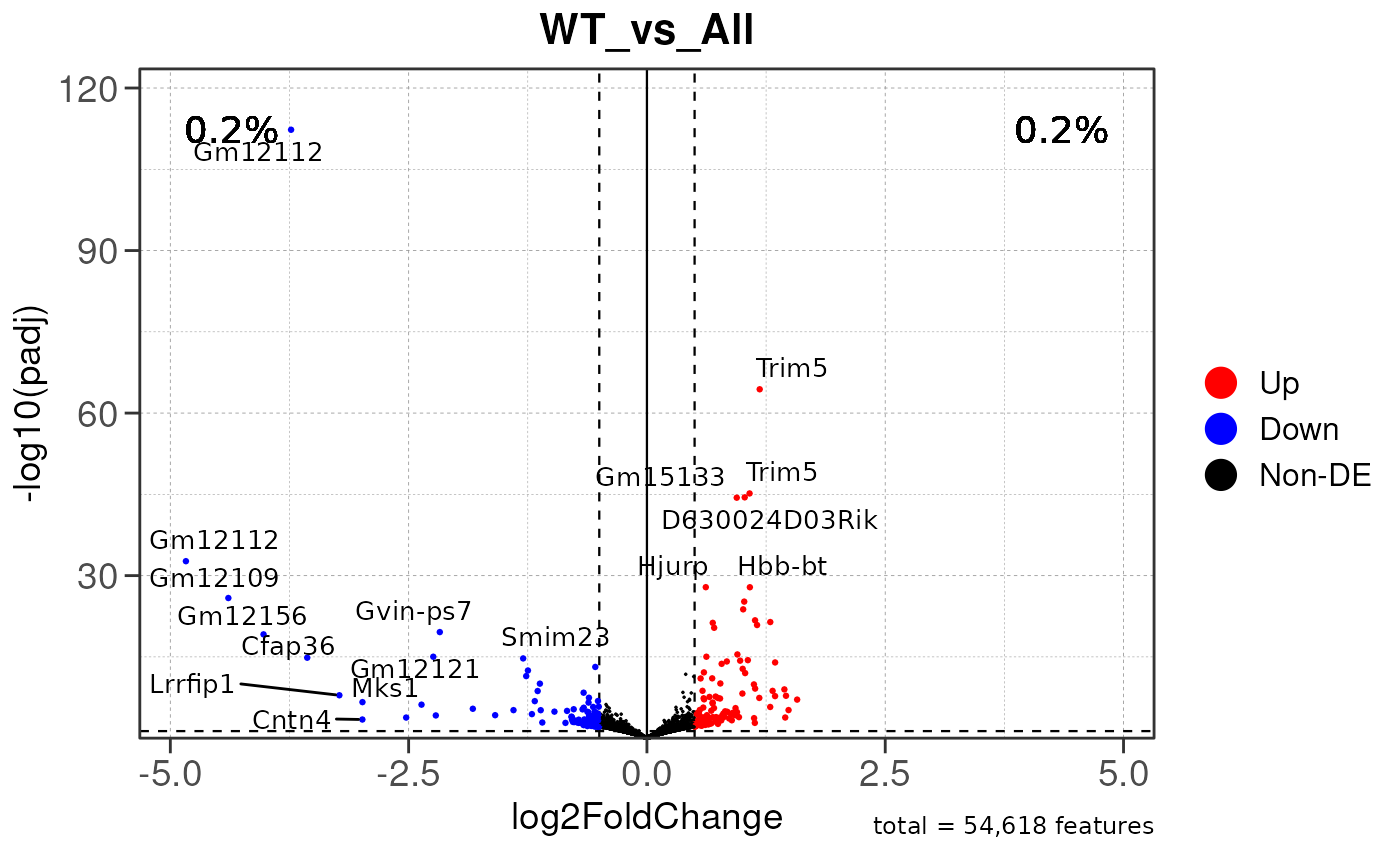
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## -- replacing outliers and refitting for 6 genes
## -- DESeq argument 'minReplicatesForReplace' = 7
## -- original counts are preserved in counts(dds)
## estimating dispersions
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 768 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 768 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 59222 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 16 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 59222 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 22 unlabeled data points (too many overlaps). Consider increasing max.overlaps
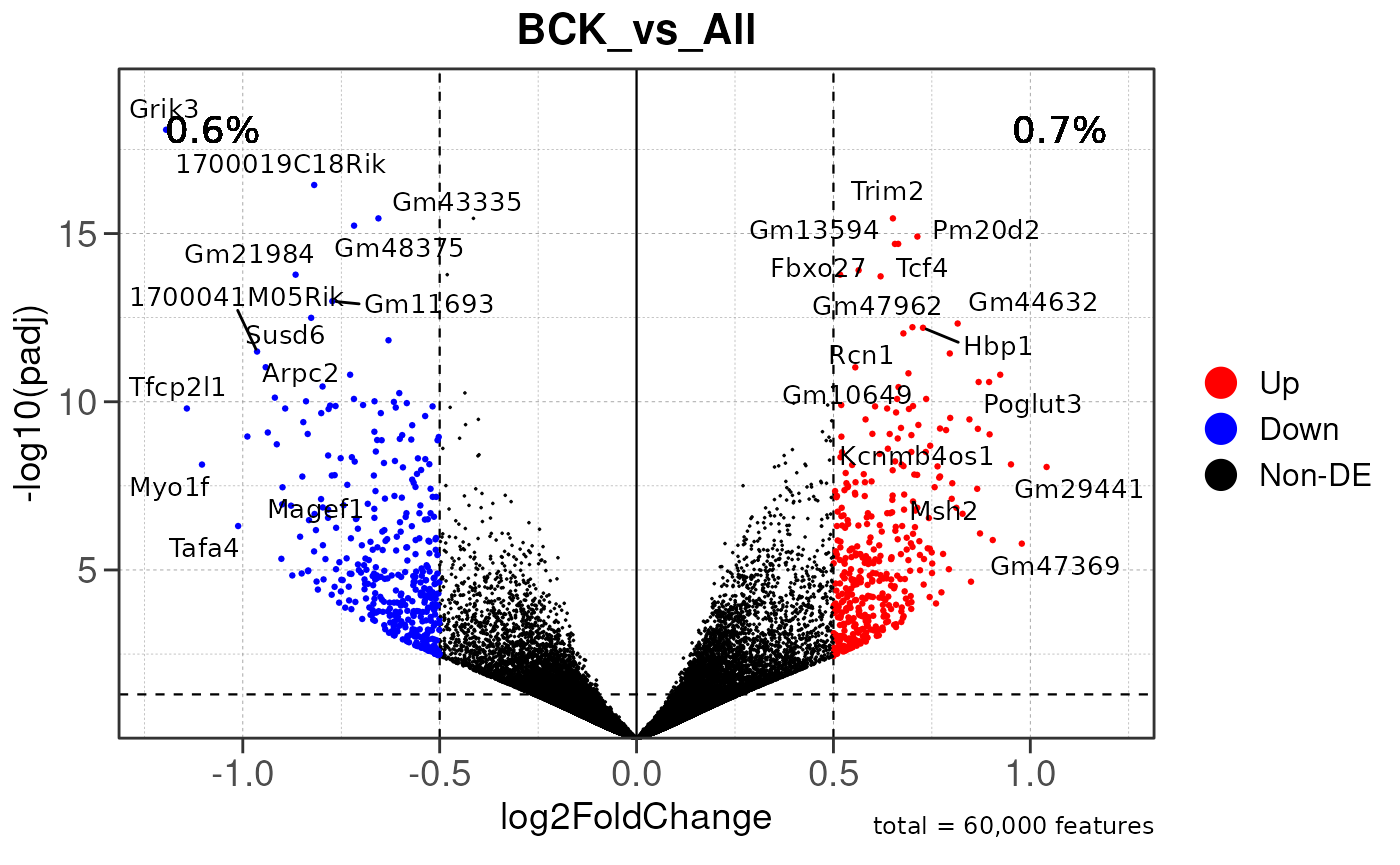
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## -- replacing outliers and refitting for 6 genes
## -- DESeq argument 'minReplicatesForReplace' = 7
## -- original counts are preserved in counts(dds)
## estimating dispersions
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 9 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 61343 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 61343 rows containing missing values or values outside the scale range
## (`geom_text()`).
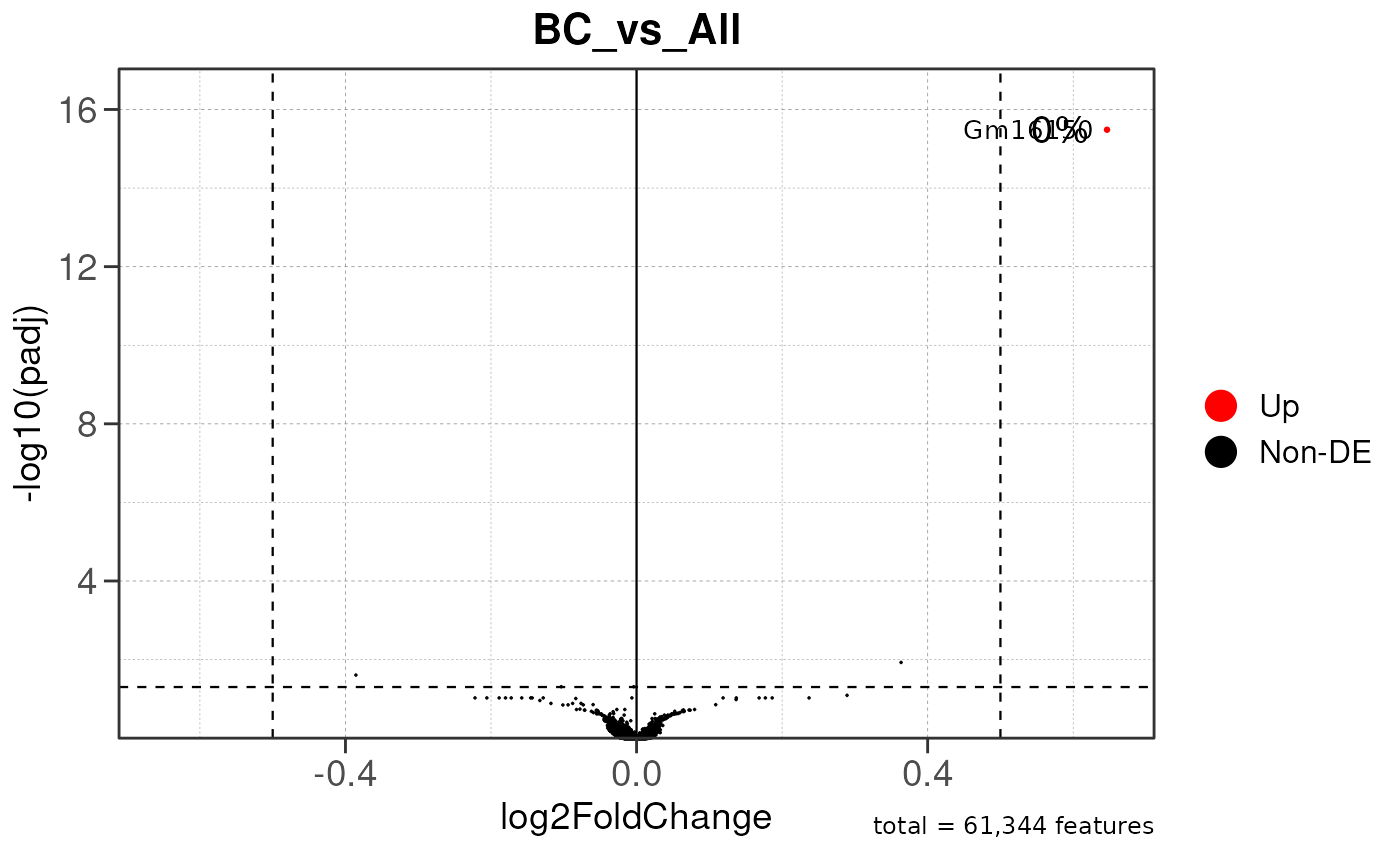
## Generating ONE-TO-ALL peak annotation plots for ALL PEAKS...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 241 peaks in All DE Peaks...
## Creating pie charts for 122 peaks in Upregulated DE Peaks...
## Creating pie charts for 119 peaks in Downregulated DE Peaks...
## Saving pie charts to PDF...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 778 peaks in All DE Peaks...
## Creating pie charts for 395 peaks in Upregulated DE Peaks...
## Creating pie charts for 383 peaks in Downregulated DE Peaks...
## Saving pie charts to PDF...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Filtering differentially expressed peaks...
## Processing peak annotations...
## Plotting pie charts...
## Creating pie charts for 1 peaks in All DE Peaks...
## Creating pie charts for 1 peaks in Upregulated DE Peaks...
## Saving pie charts to PDF...
## NULL
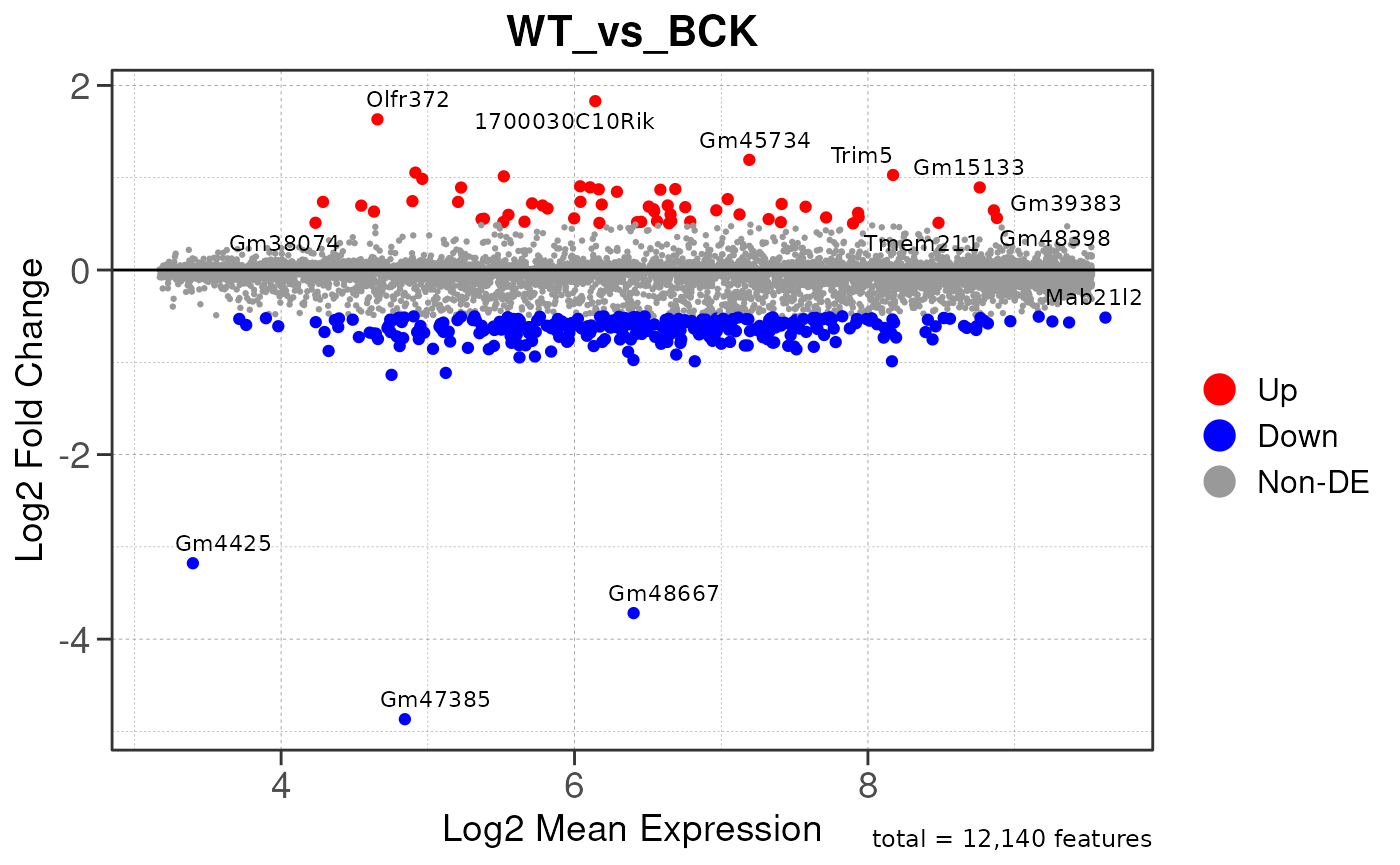
## Running PAIRWISE differential expression analysis for TSS PEAKS...
## Running PAIRWISE differential expression analysis...
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 357 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 357 unlabeled data points (too many overlaps). Consider increasing max.overlaps
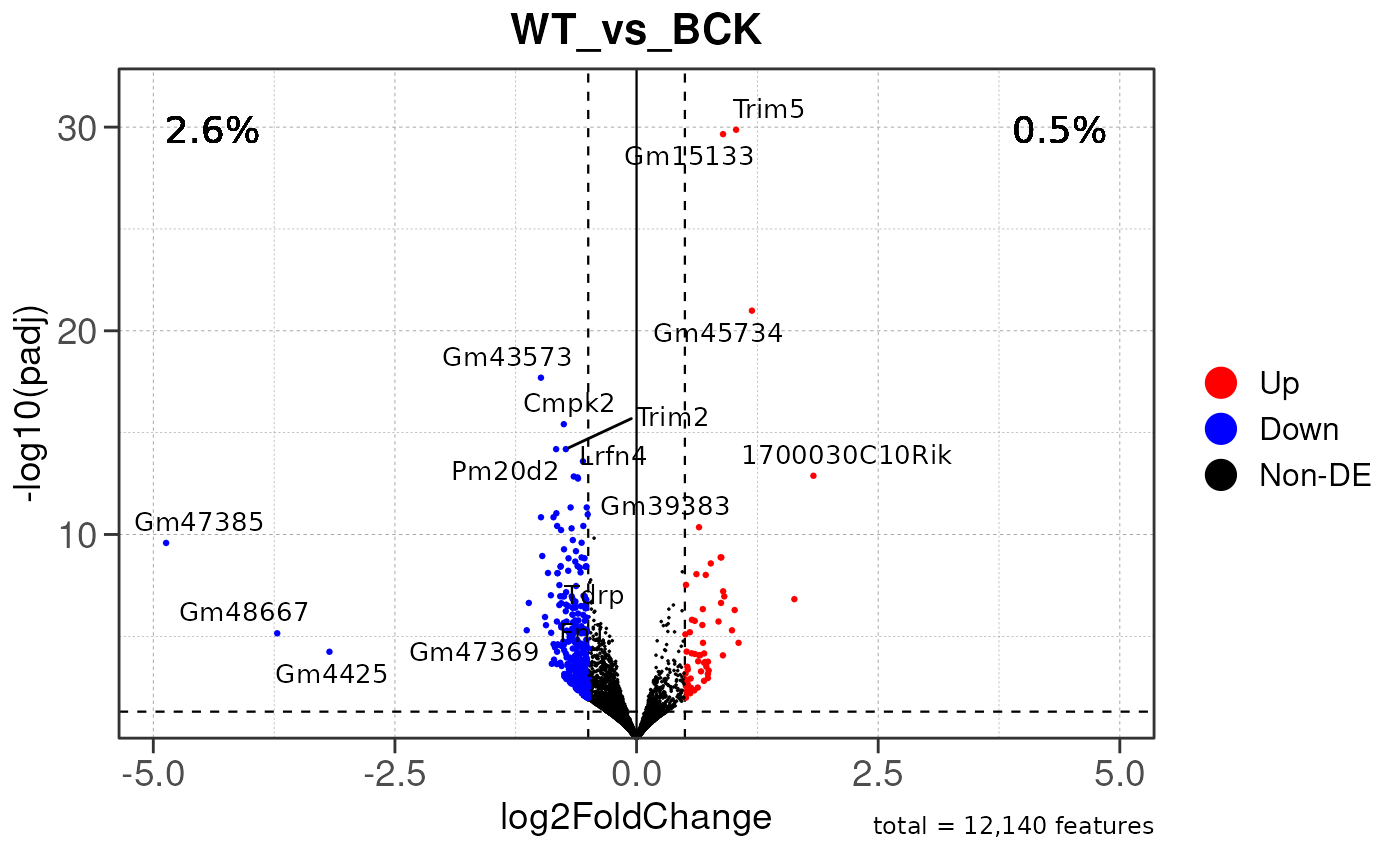
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 11770 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 30 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 11770 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 34 unlabeled data points (too many overlaps). Consider increasing max.overlaps
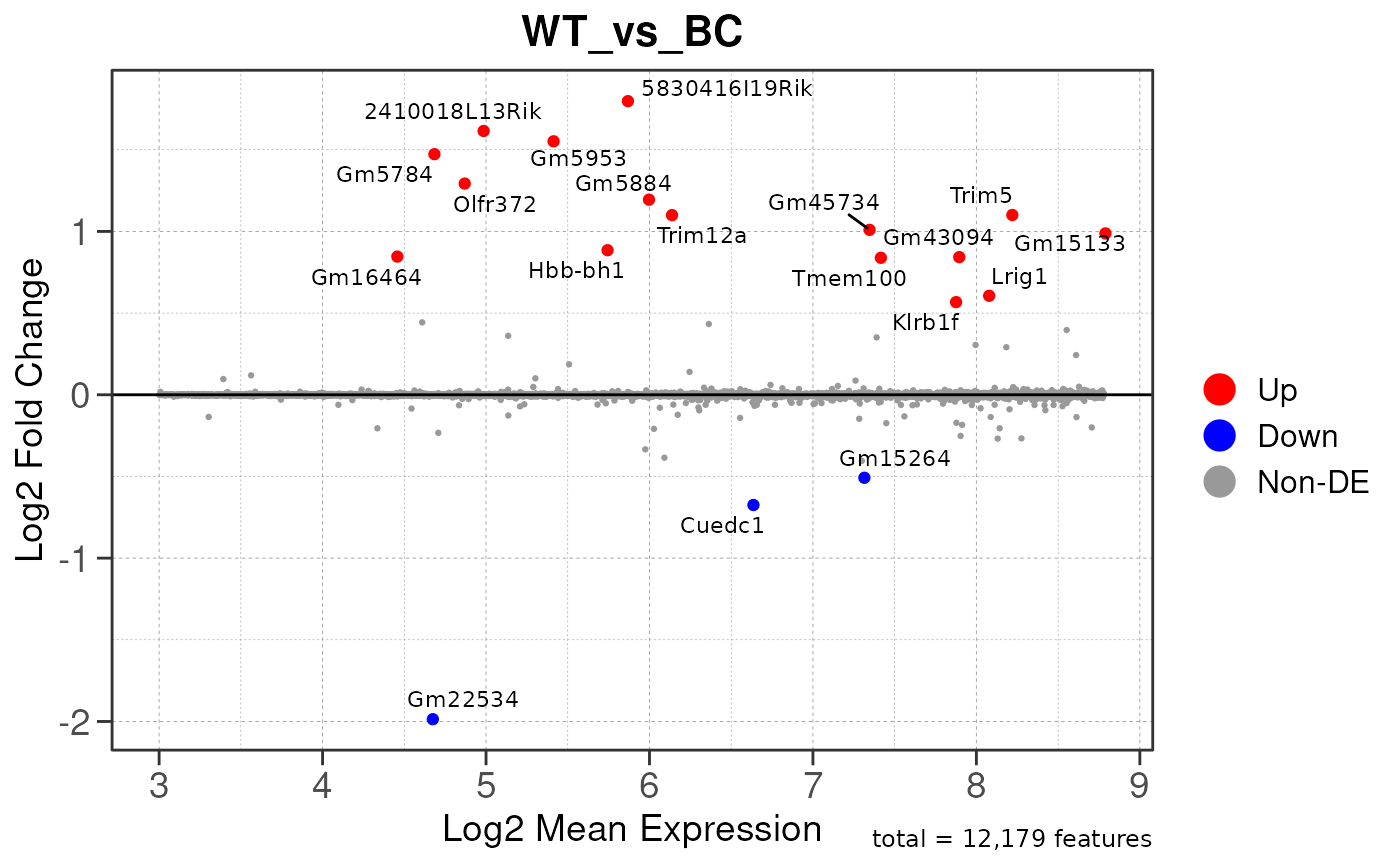
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 12360 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 12360 rows containing missing values or values outside the scale range
## (`geom_text()`).
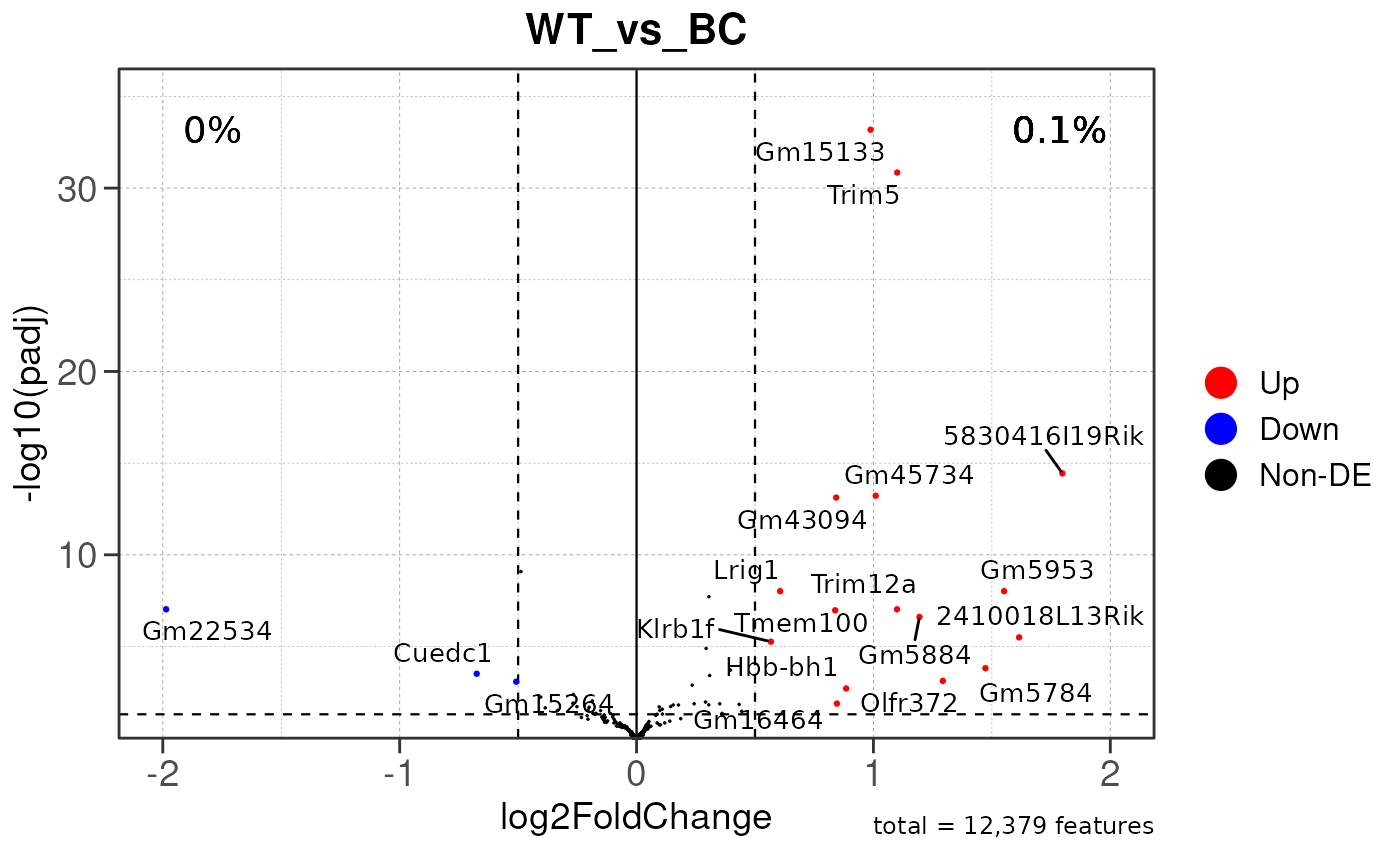
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 24 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 24 unlabeled data points (too many overlaps). Consider increasing max.overlaps
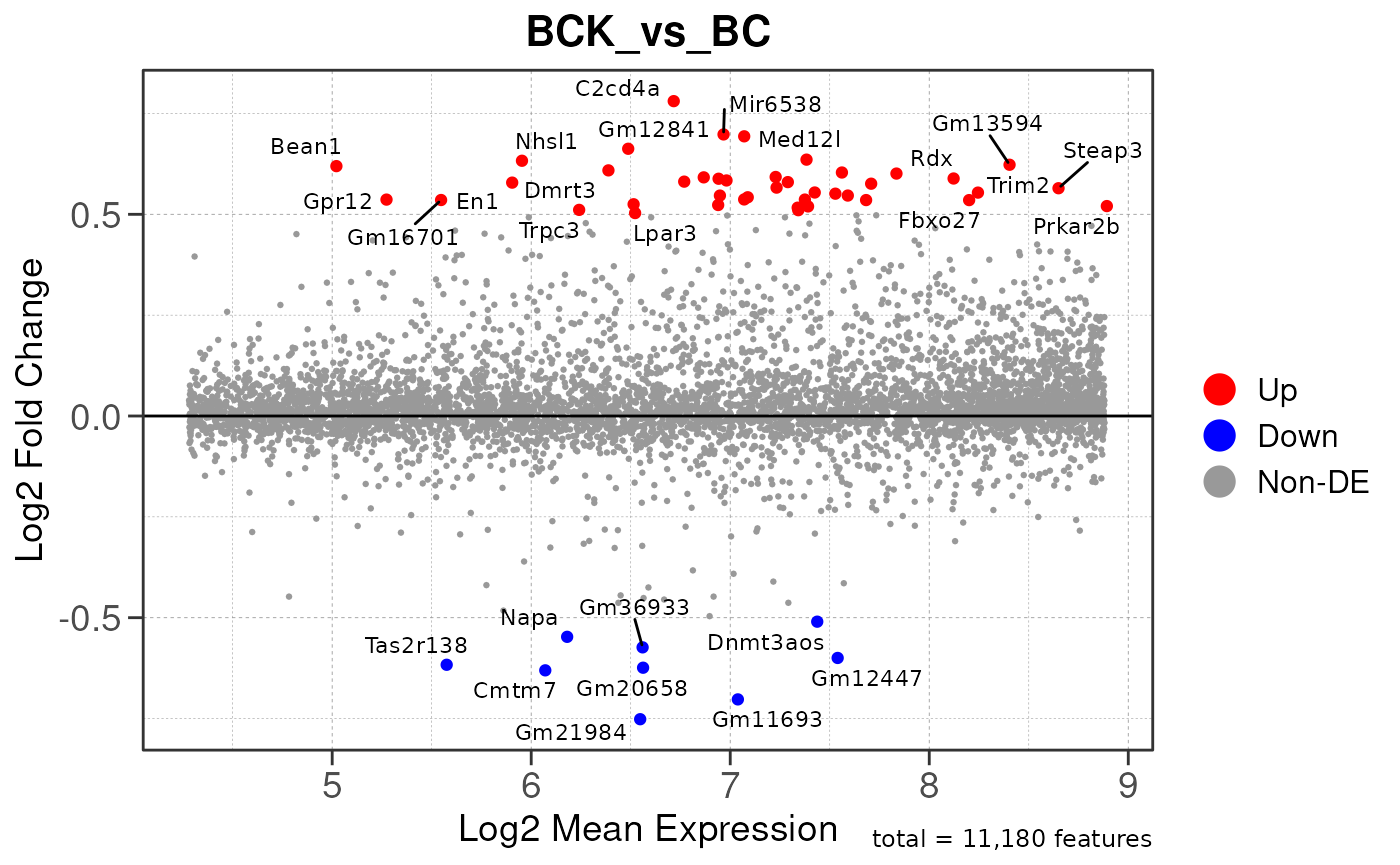
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 11129 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 3 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 11129 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 7 unlabeled data points (too many overlaps). Consider increasing max.overlaps
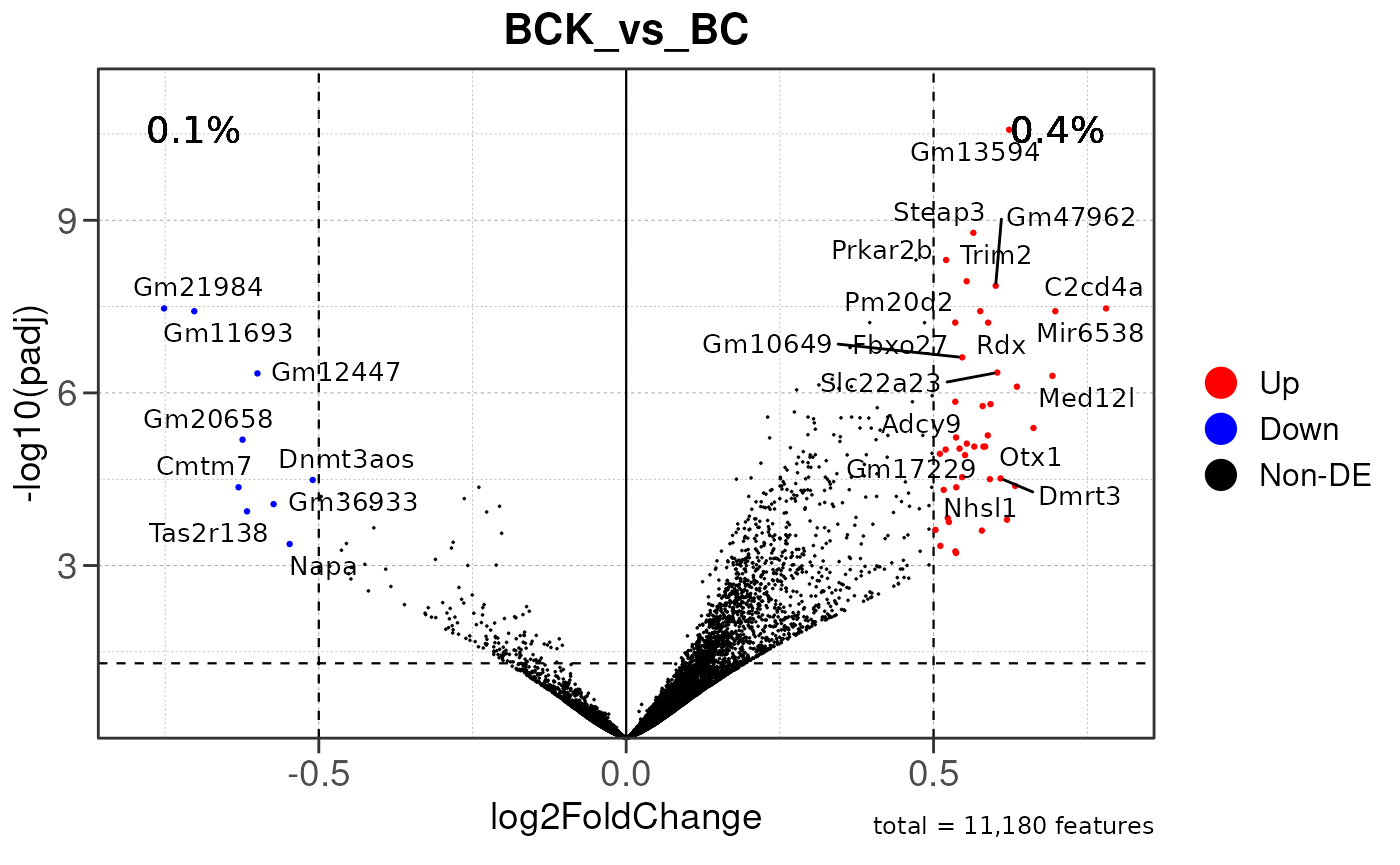
## Running PAIRWISE gene set enrichment pipeline for TSS PEAKS...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
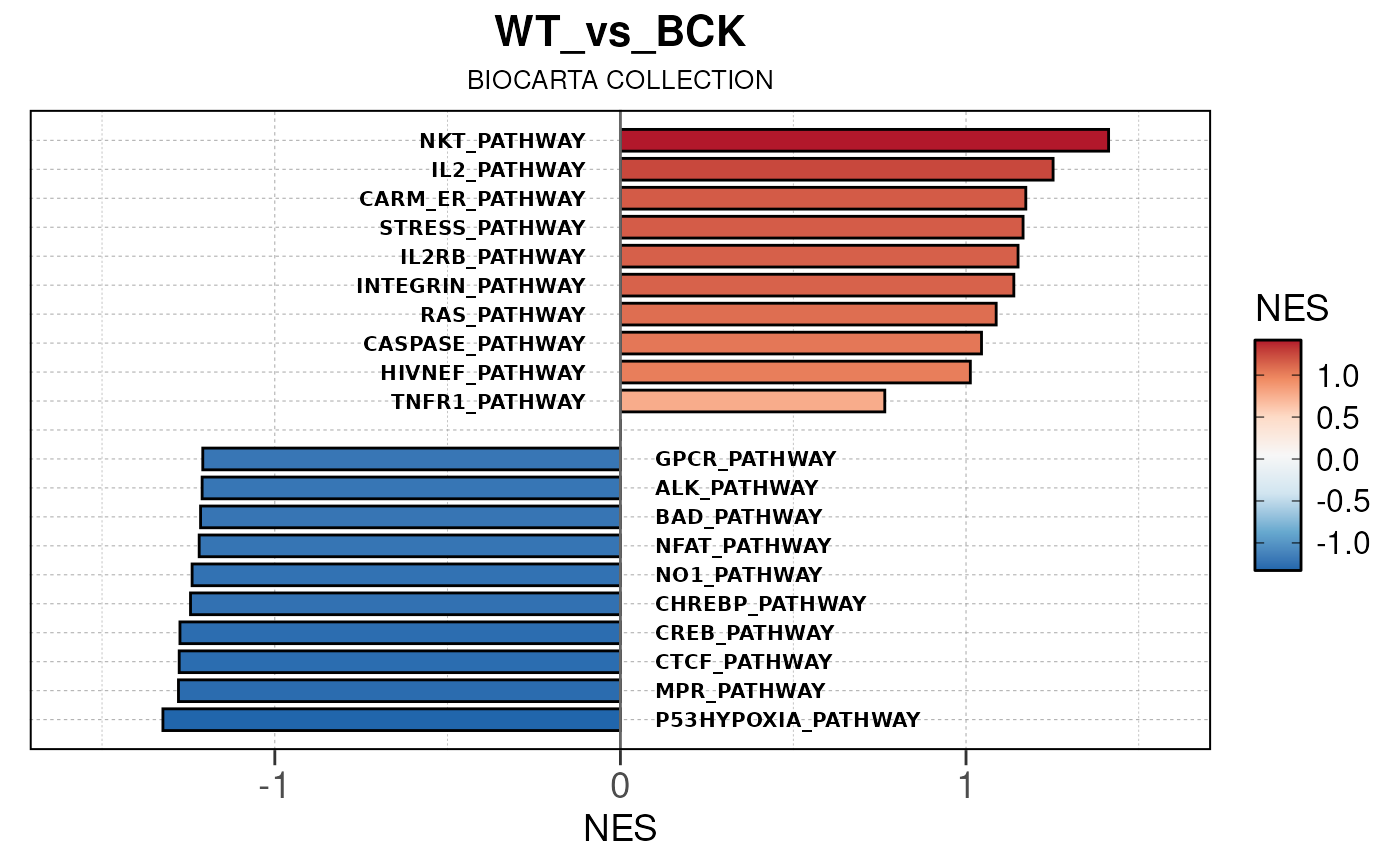
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
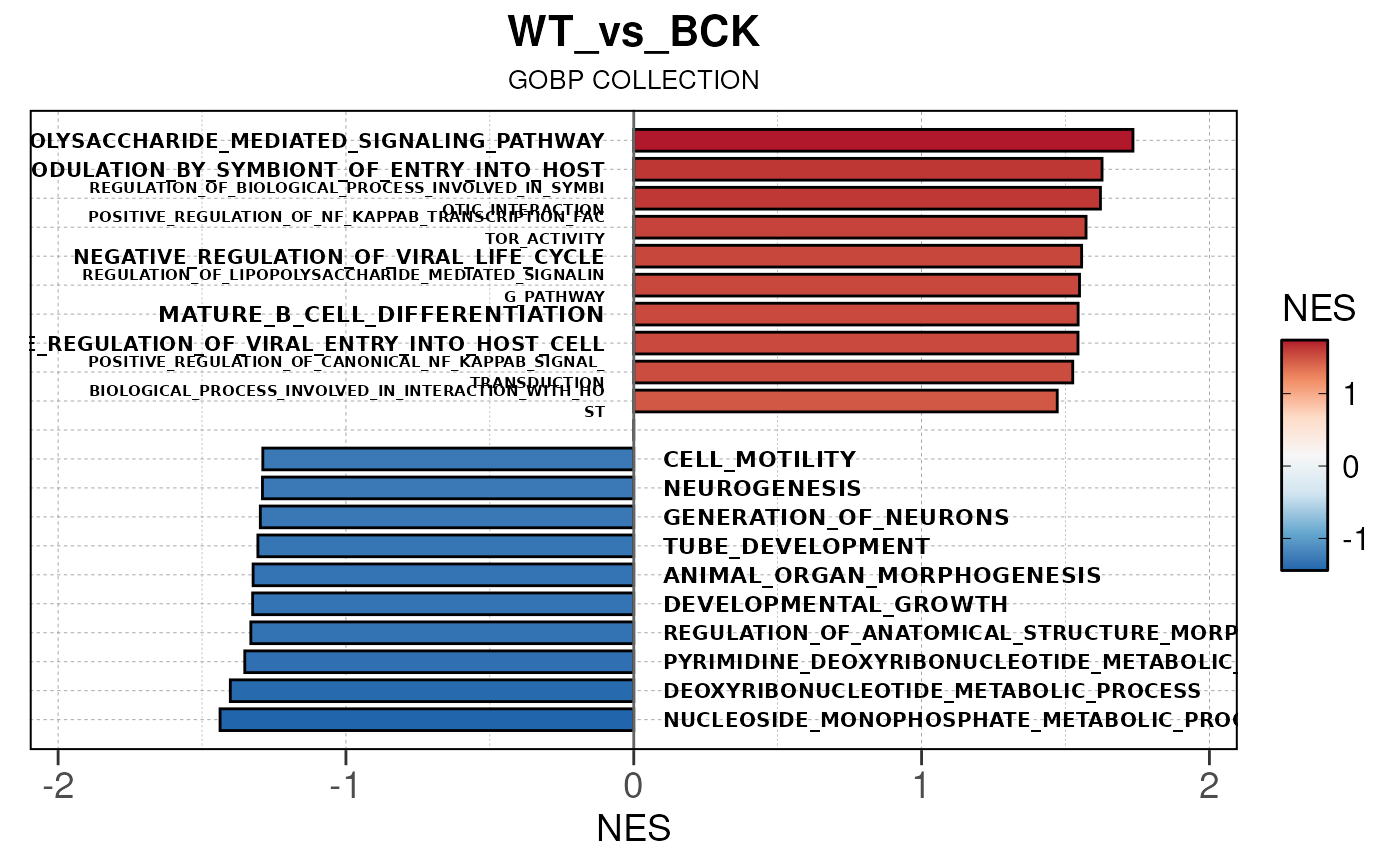
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 8 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 8 rows containing missing values or values outside the scale range
## (`geom_text()`).
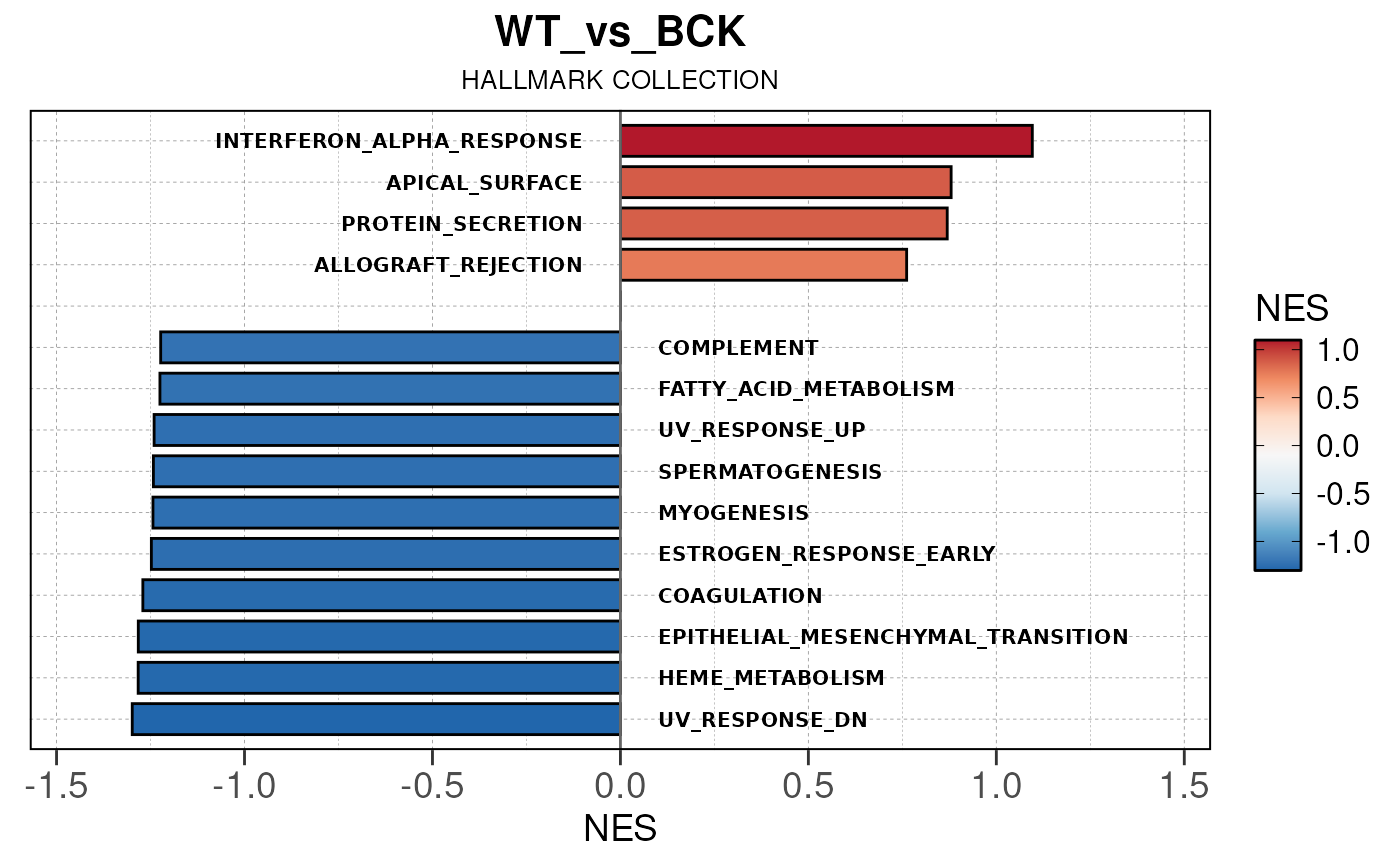
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
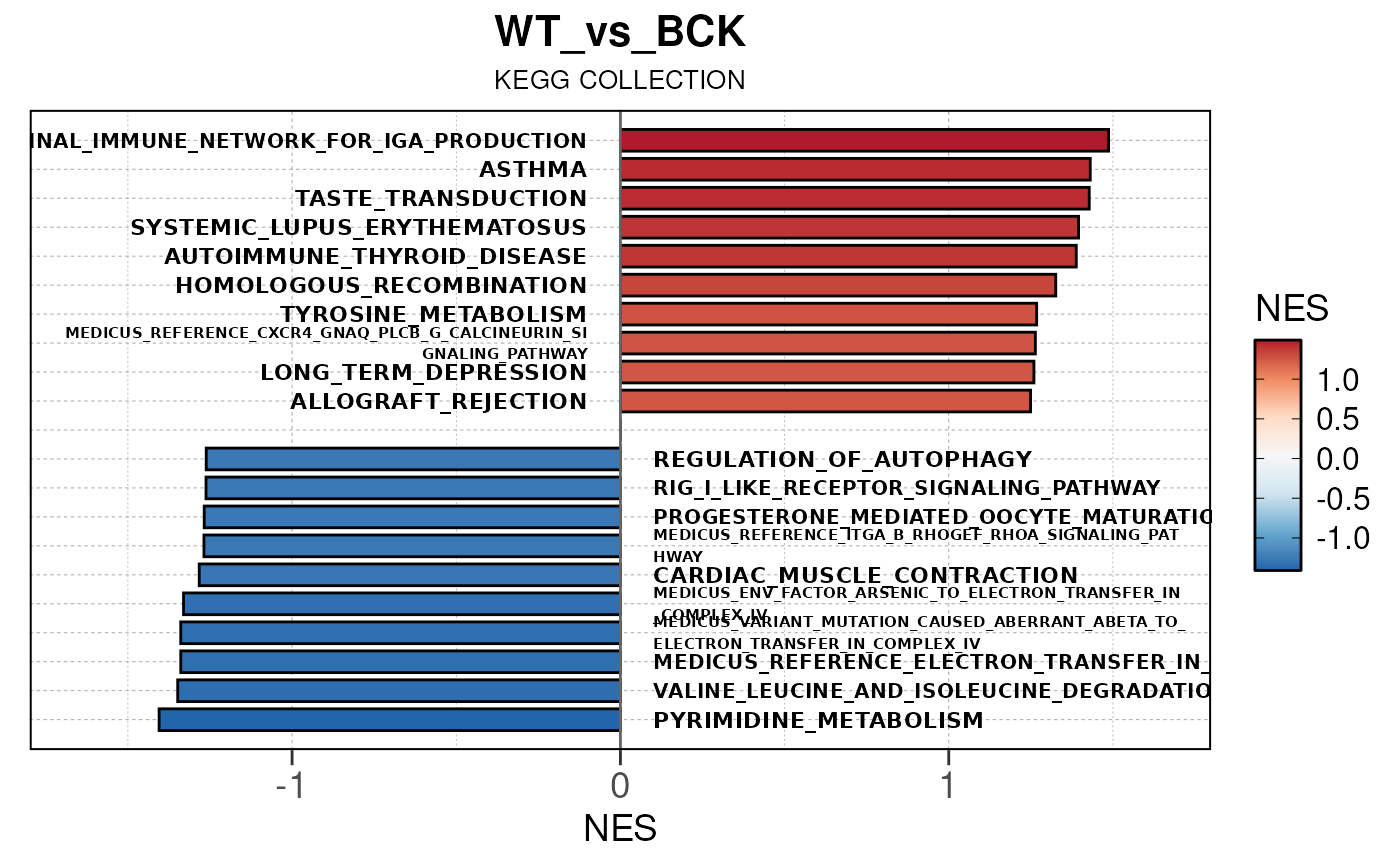
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
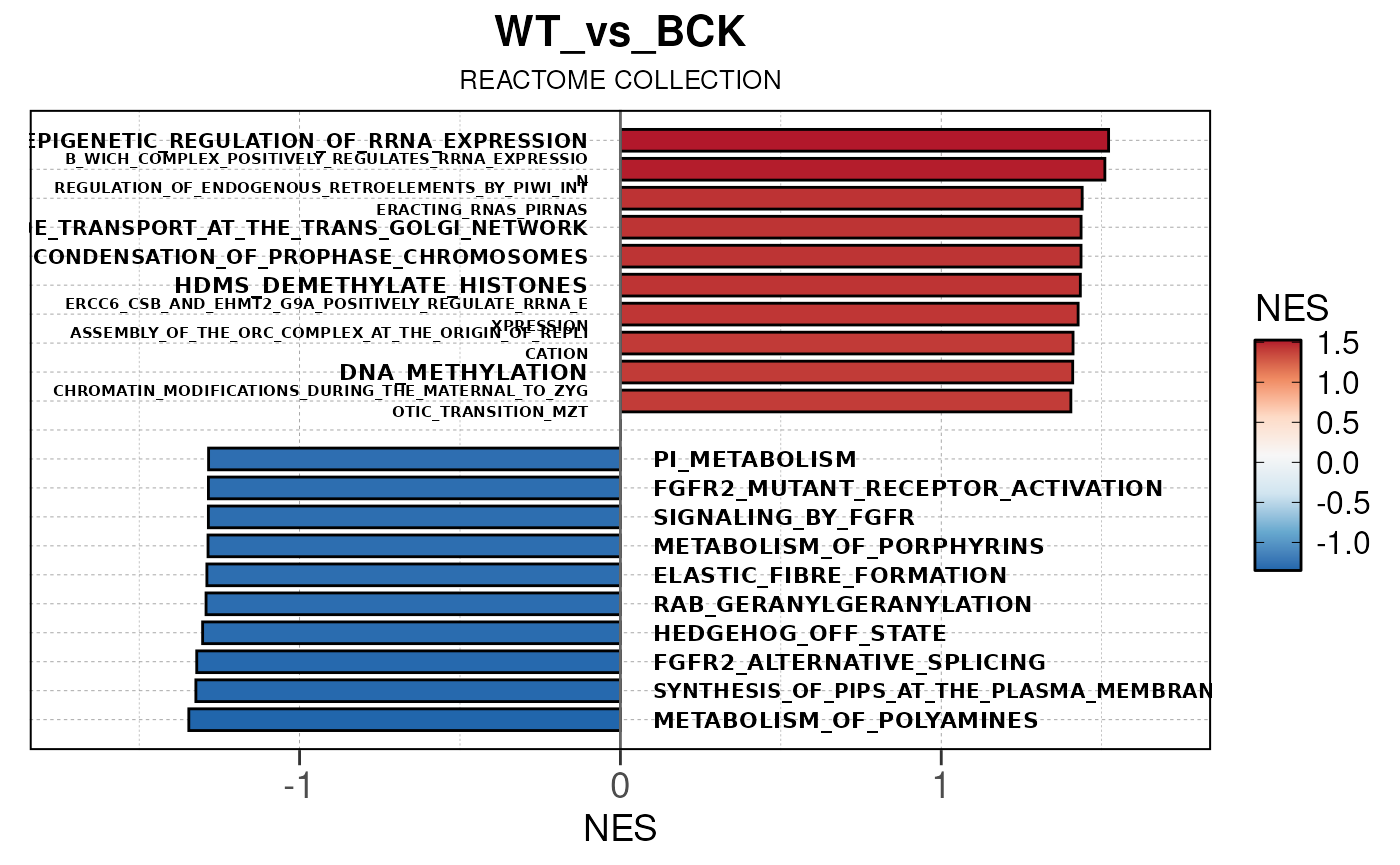
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
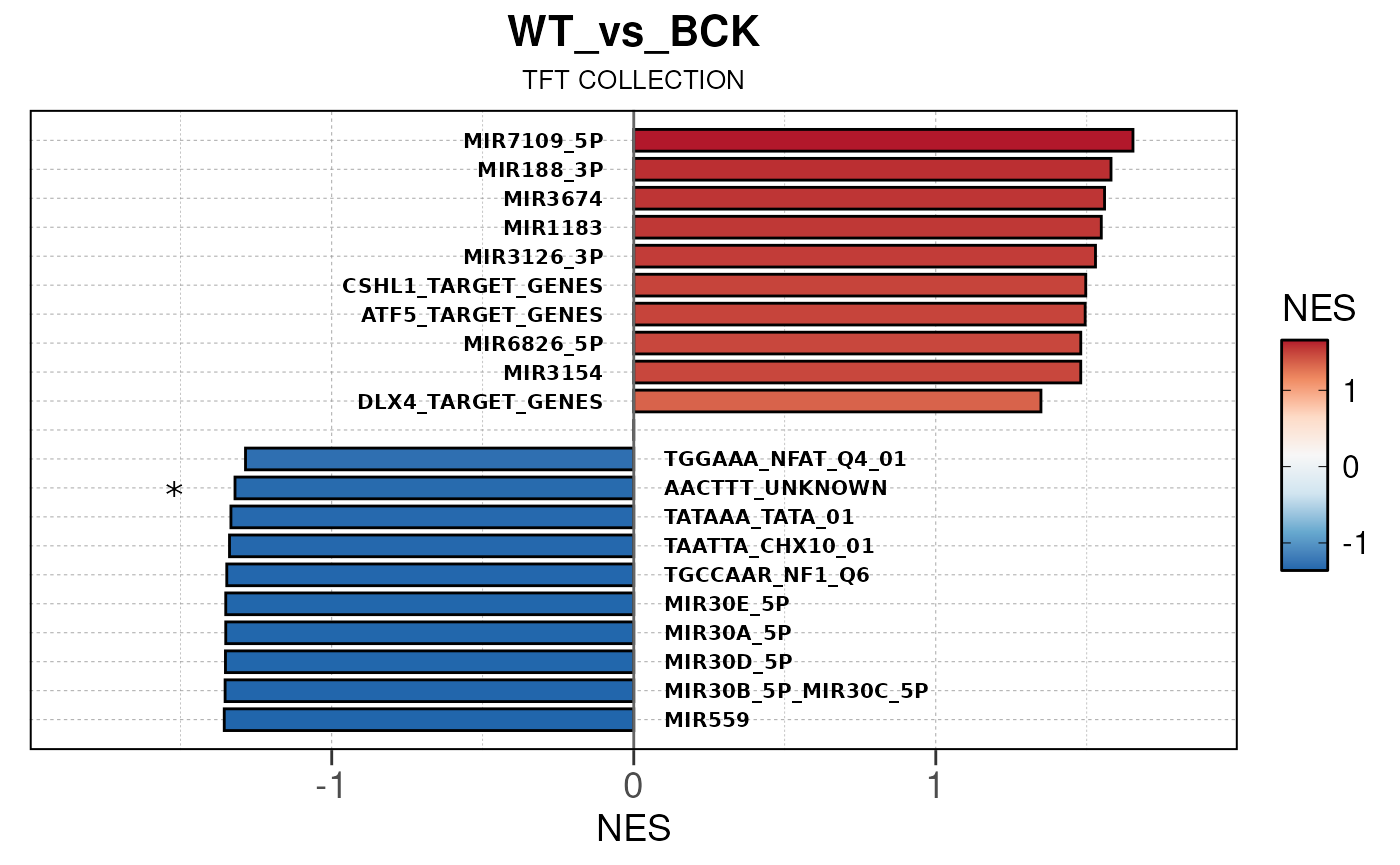
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 11 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 11 rows containing missing values or values outside the scale range
## (`geom_text()`).
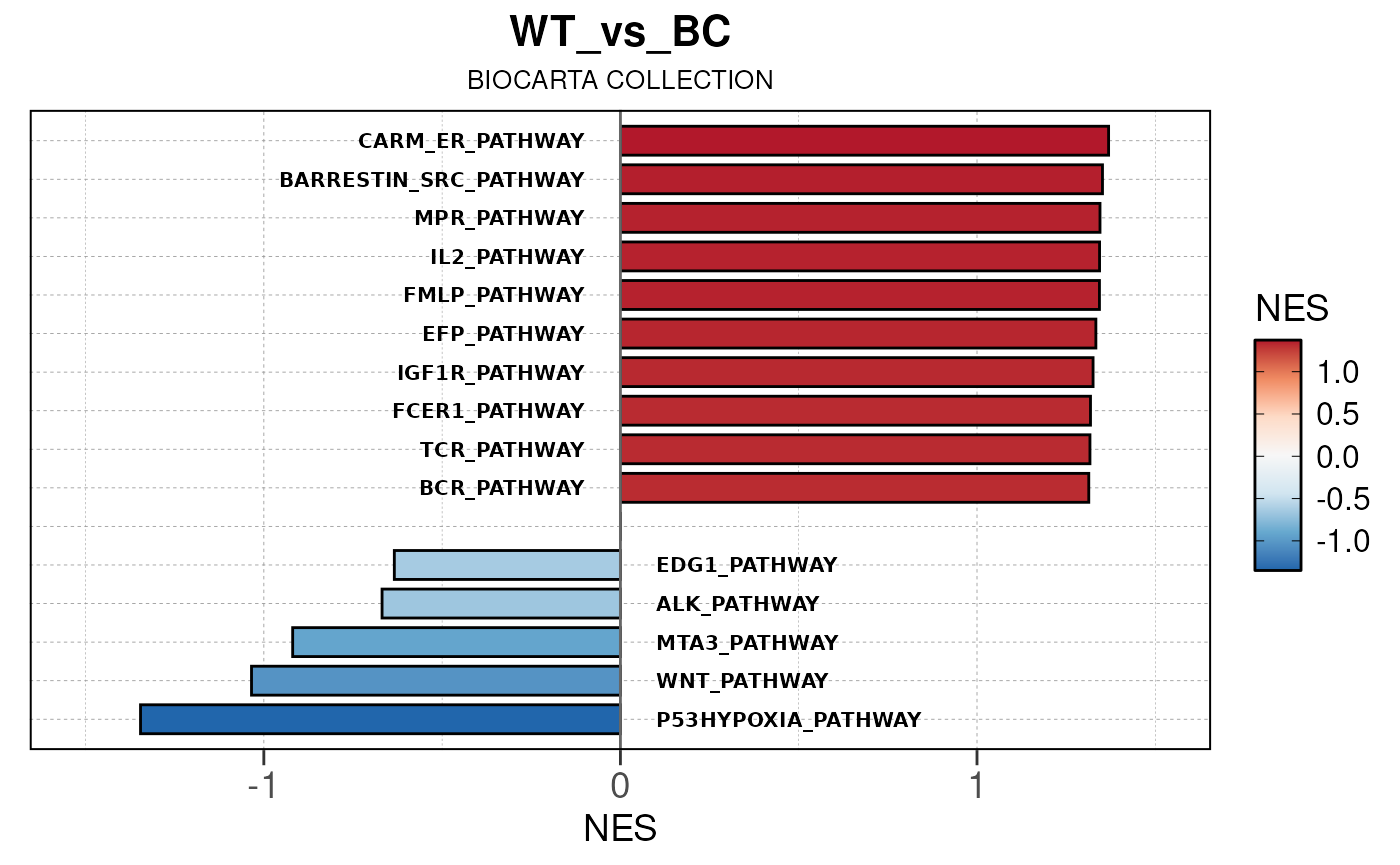
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 12 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 12 rows containing missing values or values outside the scale range
## (`geom_text()`).
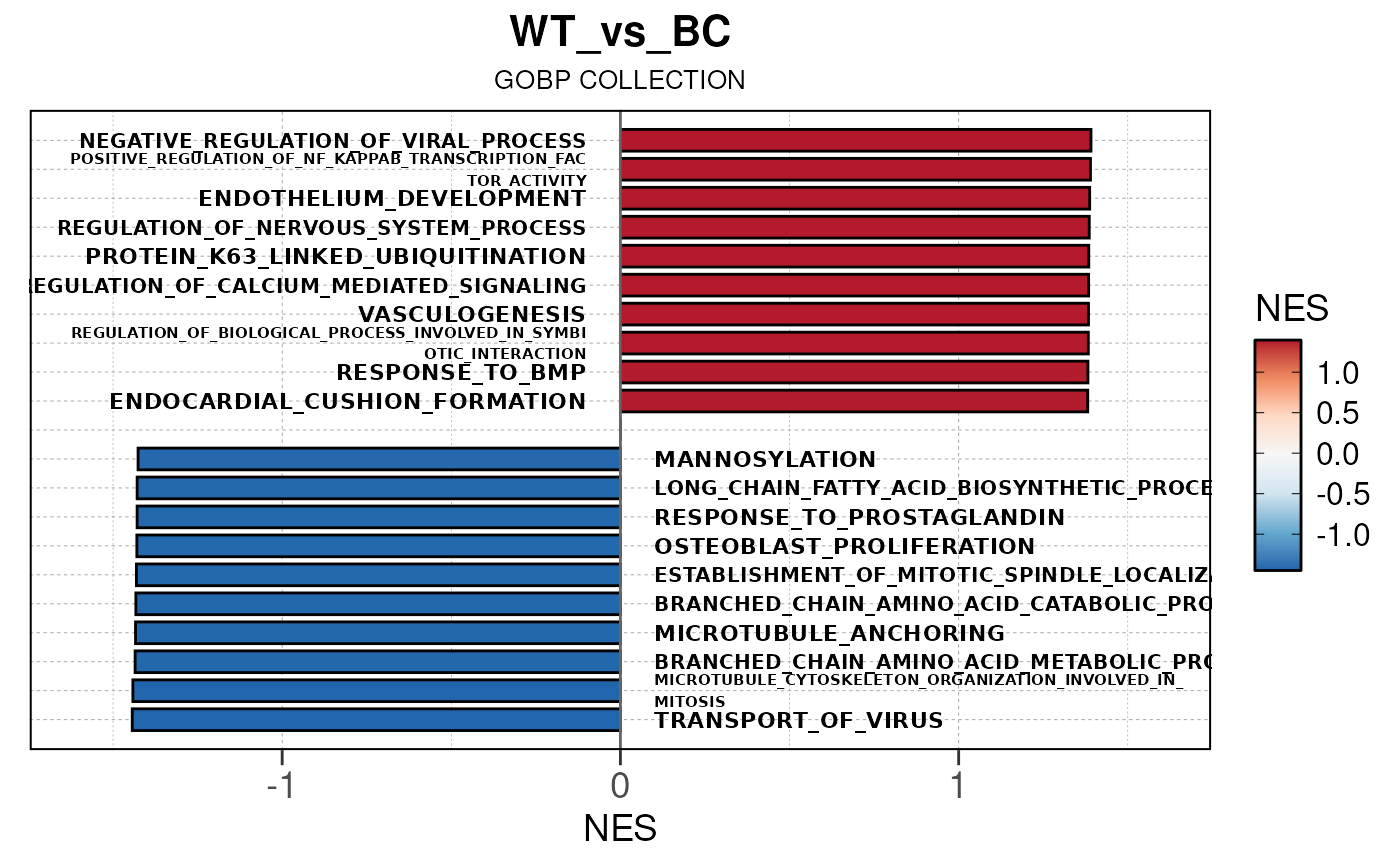
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
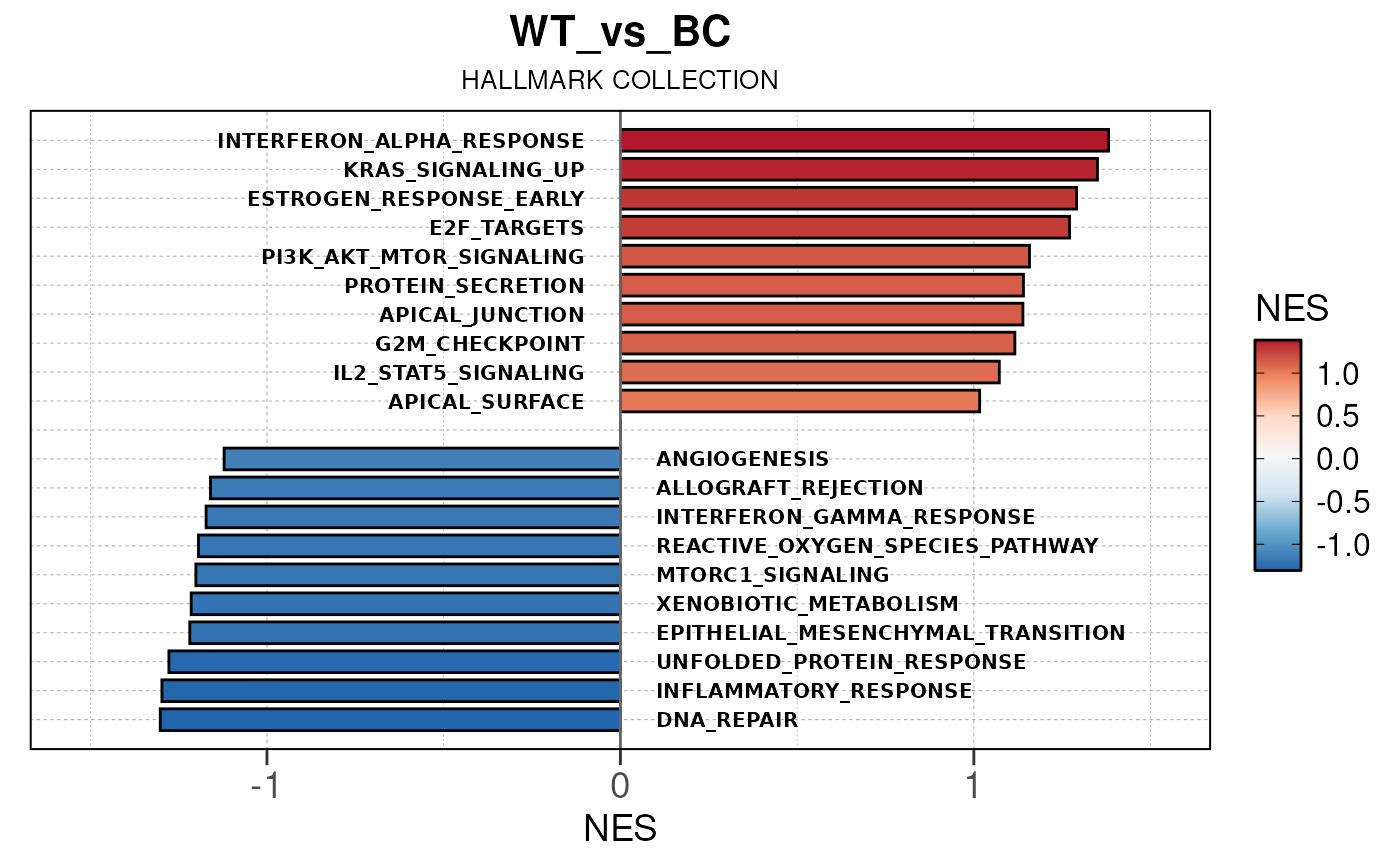
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 10 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 10 rows containing missing values or values outside the scale range
## (`geom_text()`).
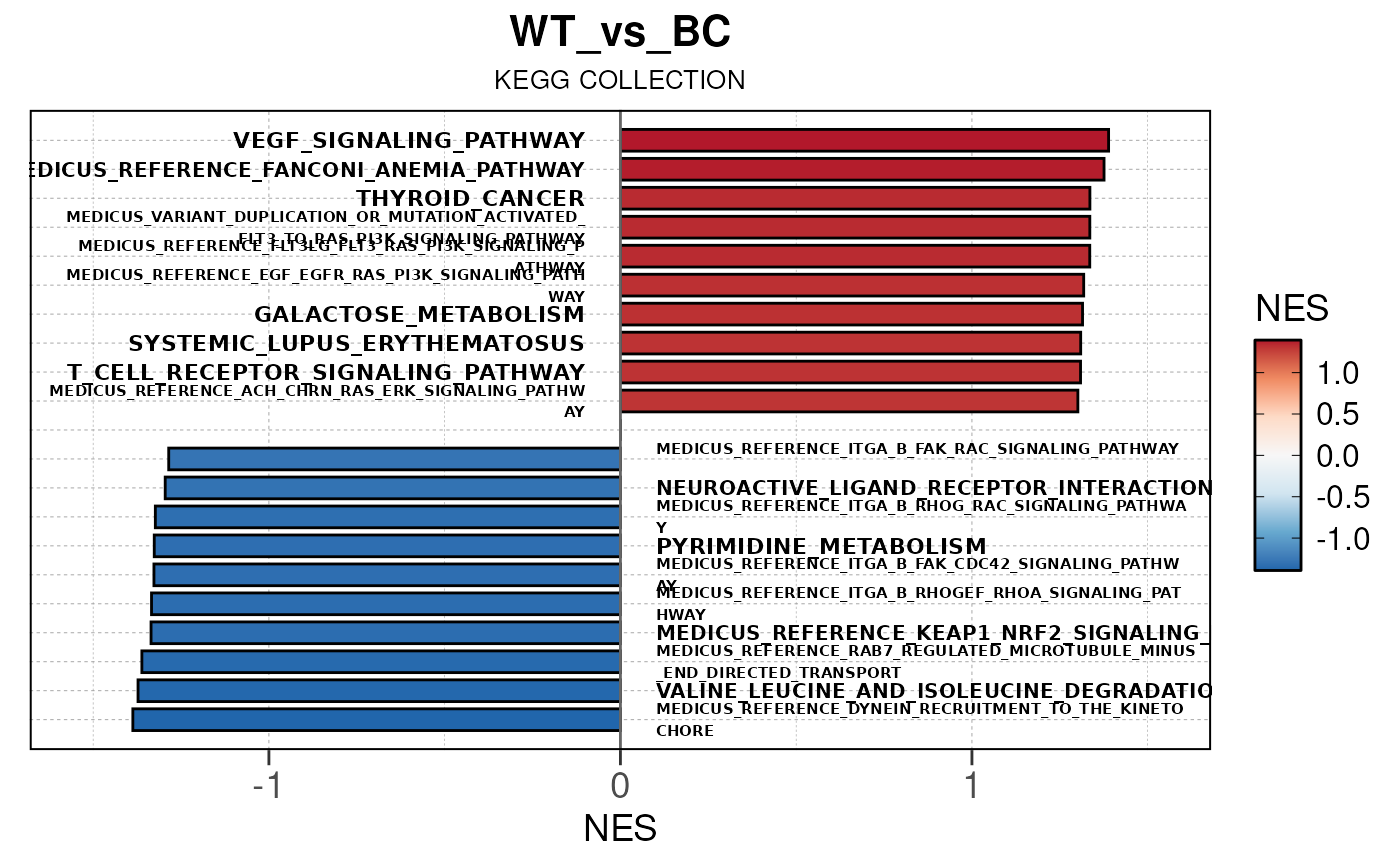
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 7 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 7 rows containing missing values or values outside the scale range
## (`geom_text()`).
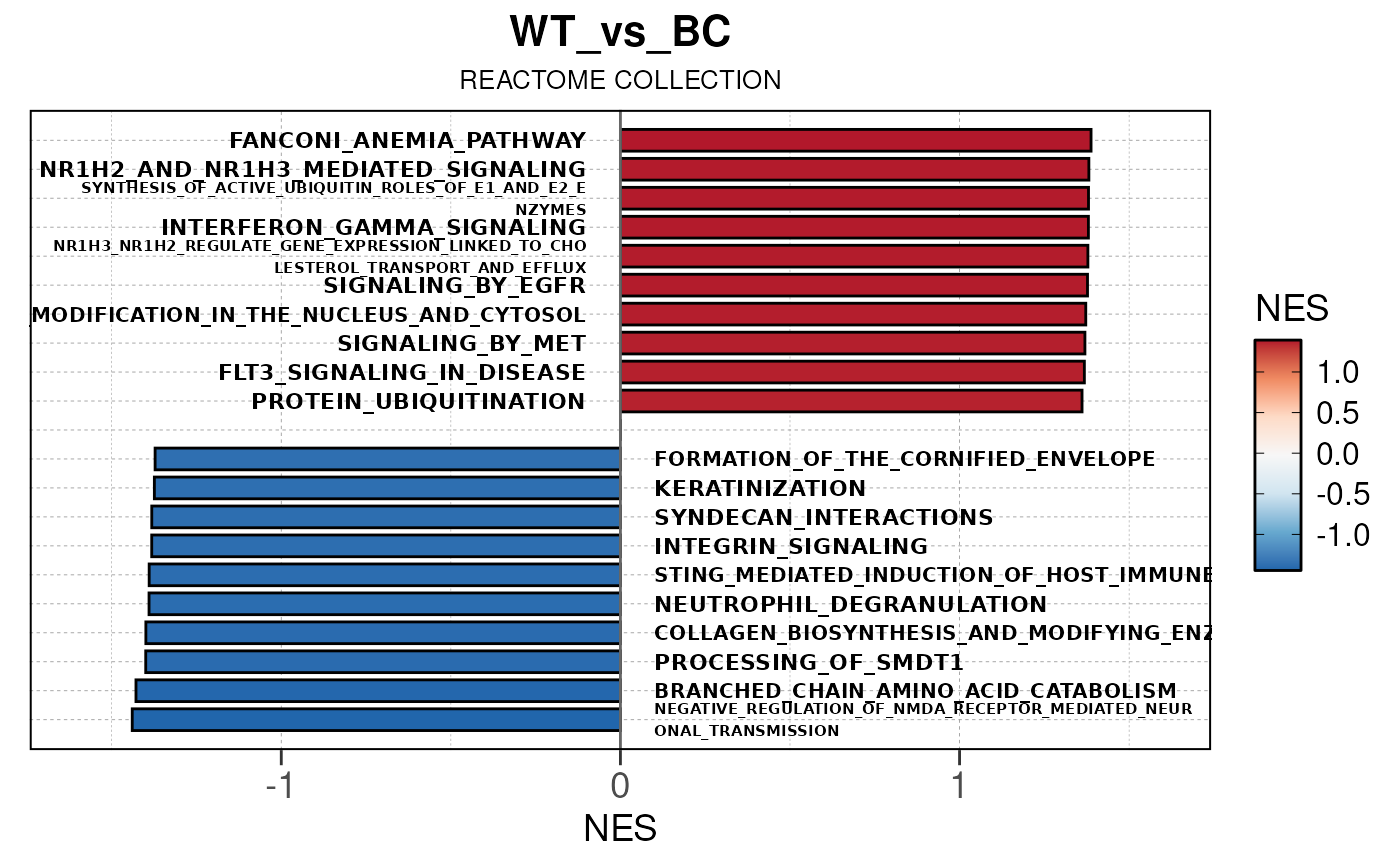
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 10 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 10 rows containing missing values or values outside the scale range
## (`geom_text()`).
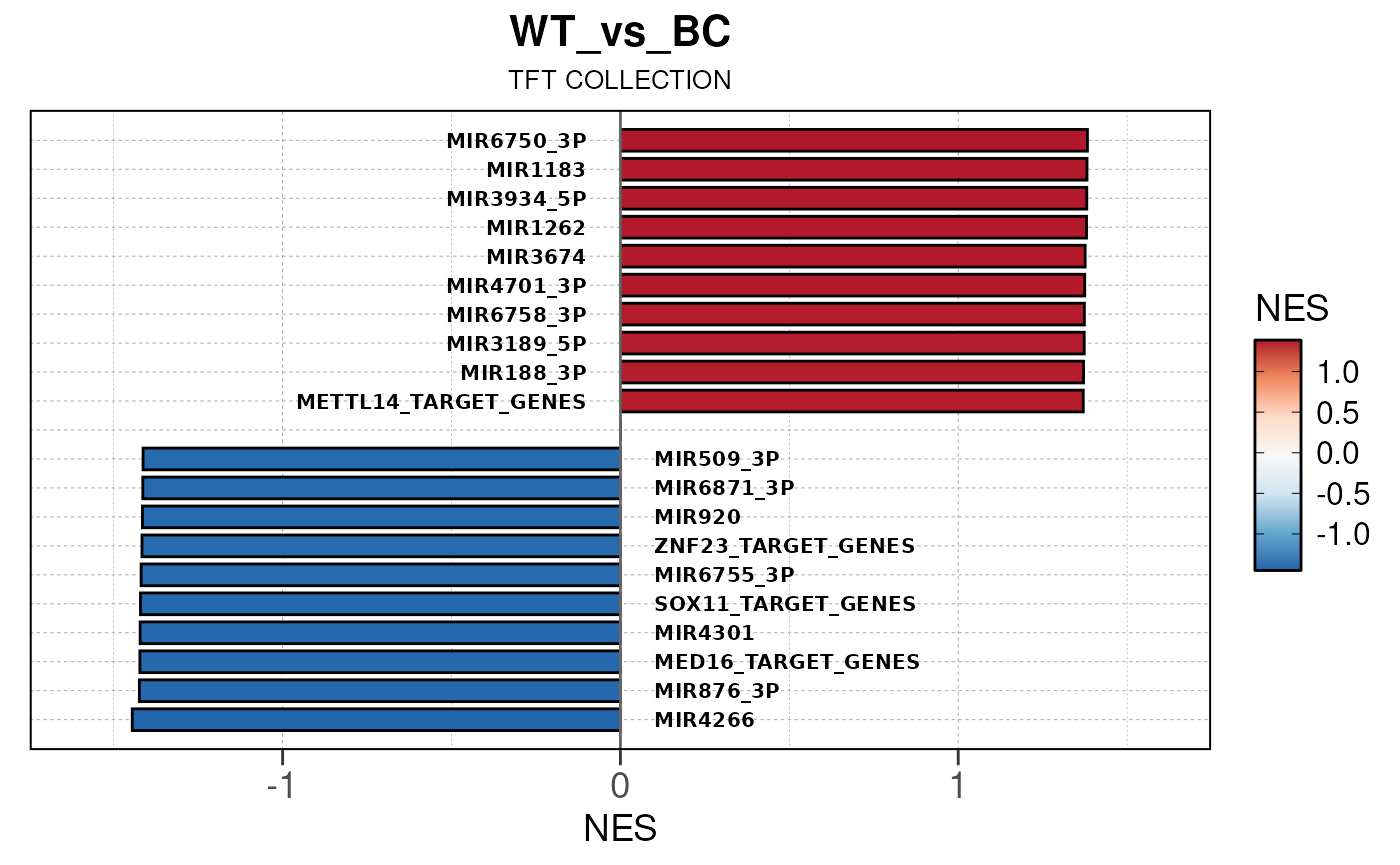
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## WARNING: There were 1 pathways for which P-values were not calculated properly due to unbalanced (positive and negative) gene-level statistic values. For such pathways pval, padj, NES, log2err are set to NA. You can try to increase the value of the argument nPermSimple (for example set it nPermSimple = 10000)
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
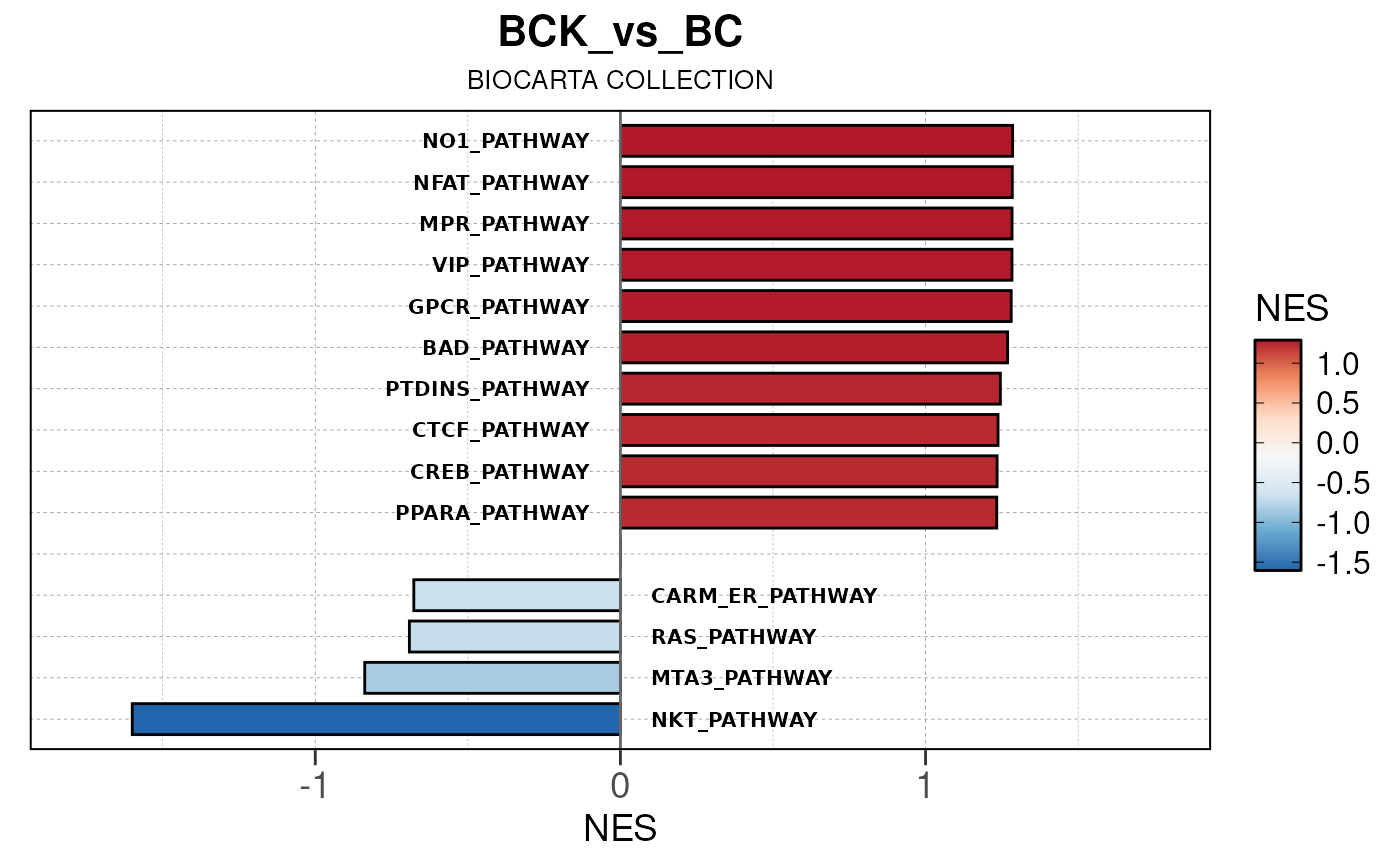
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
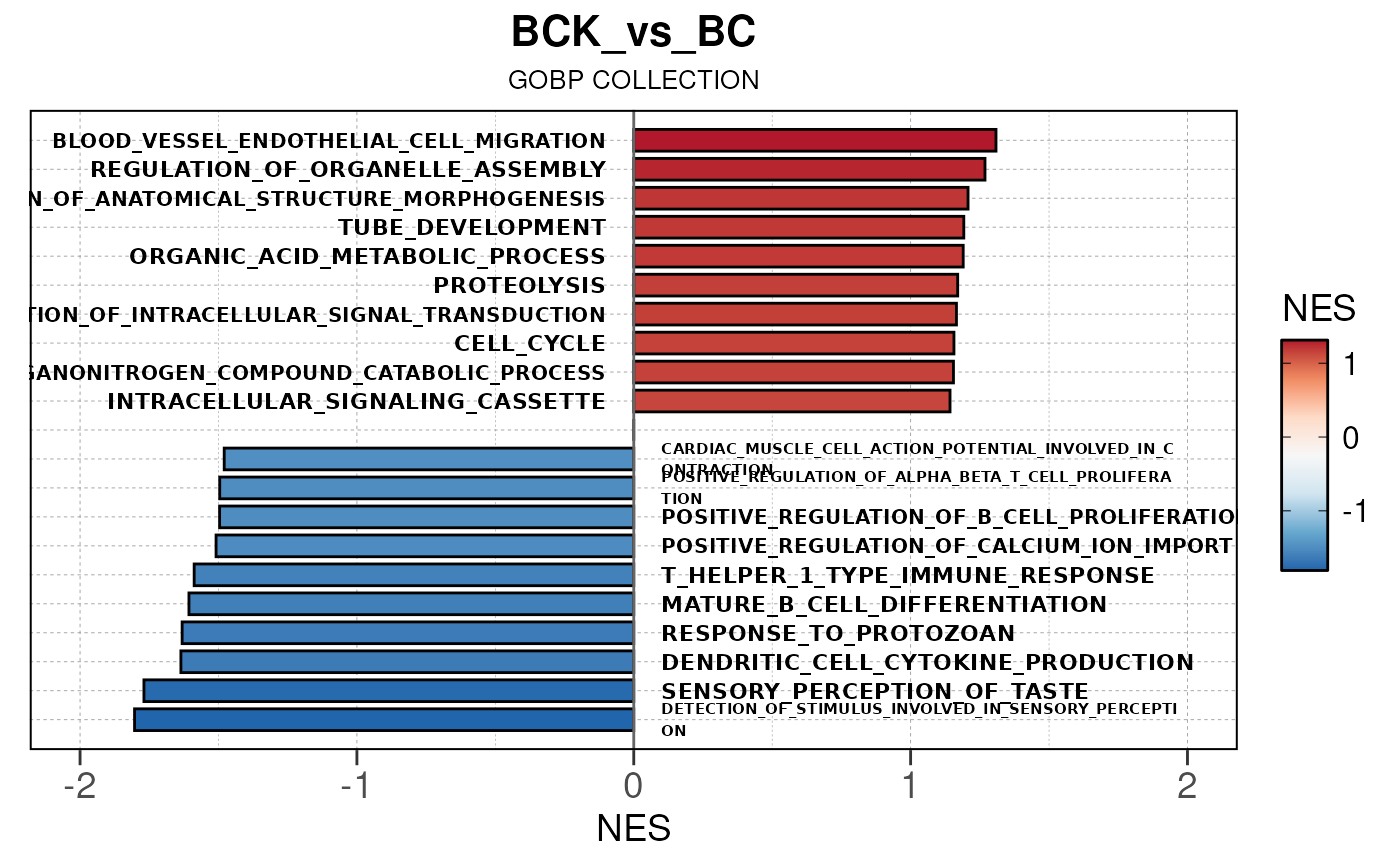
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 7 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 7 rows containing missing values or values outside the scale range
## (`geom_text()`).
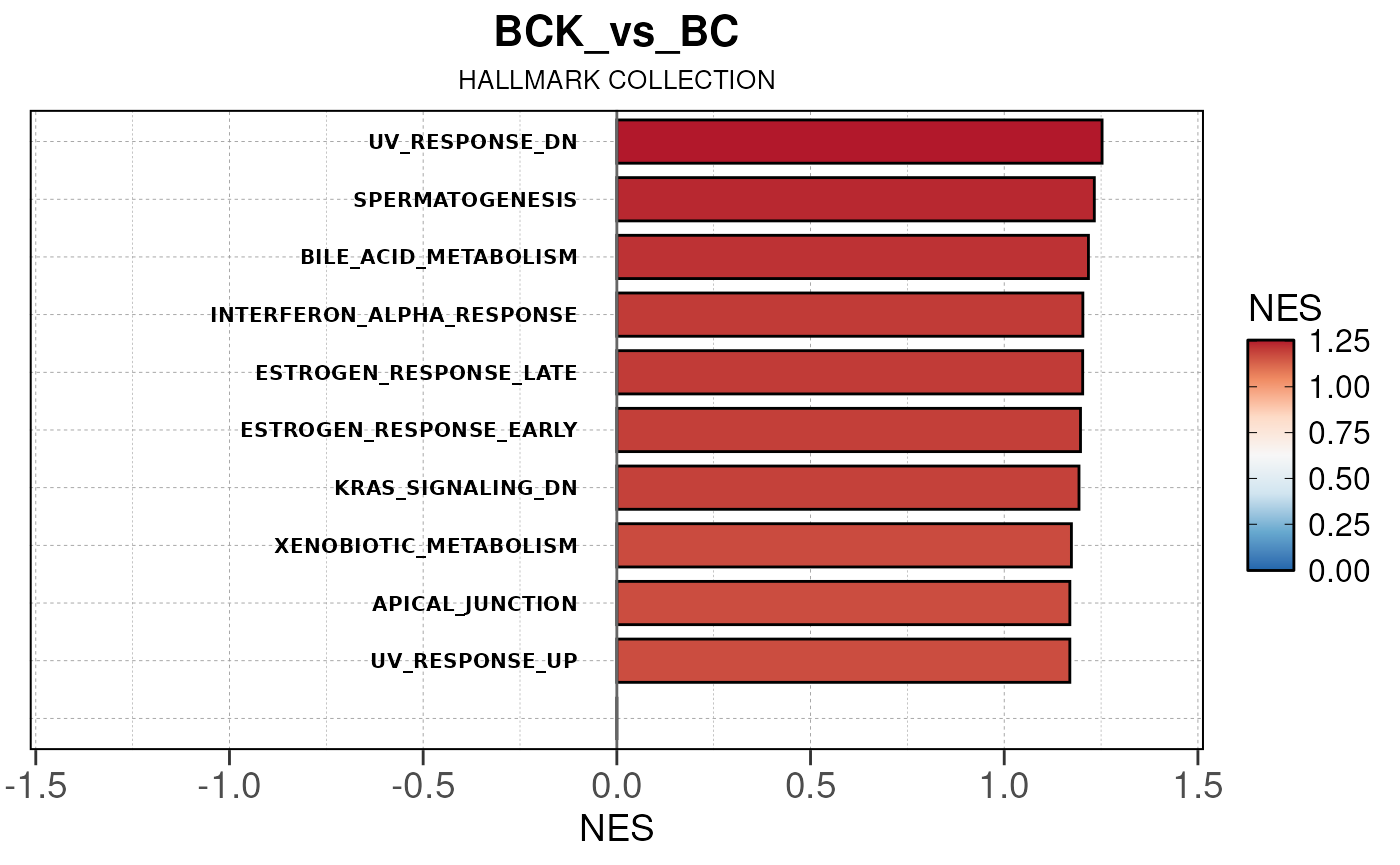
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
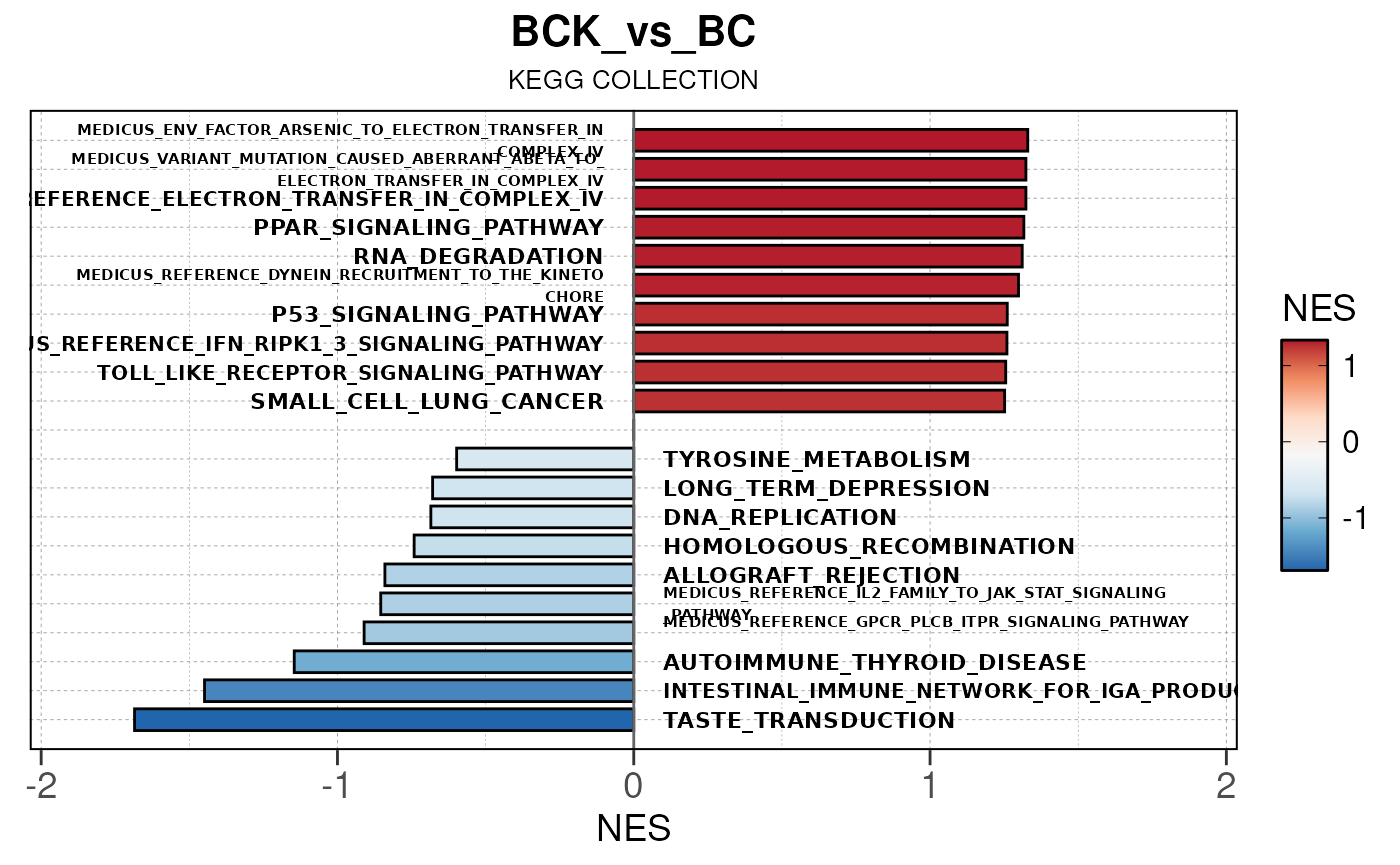
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
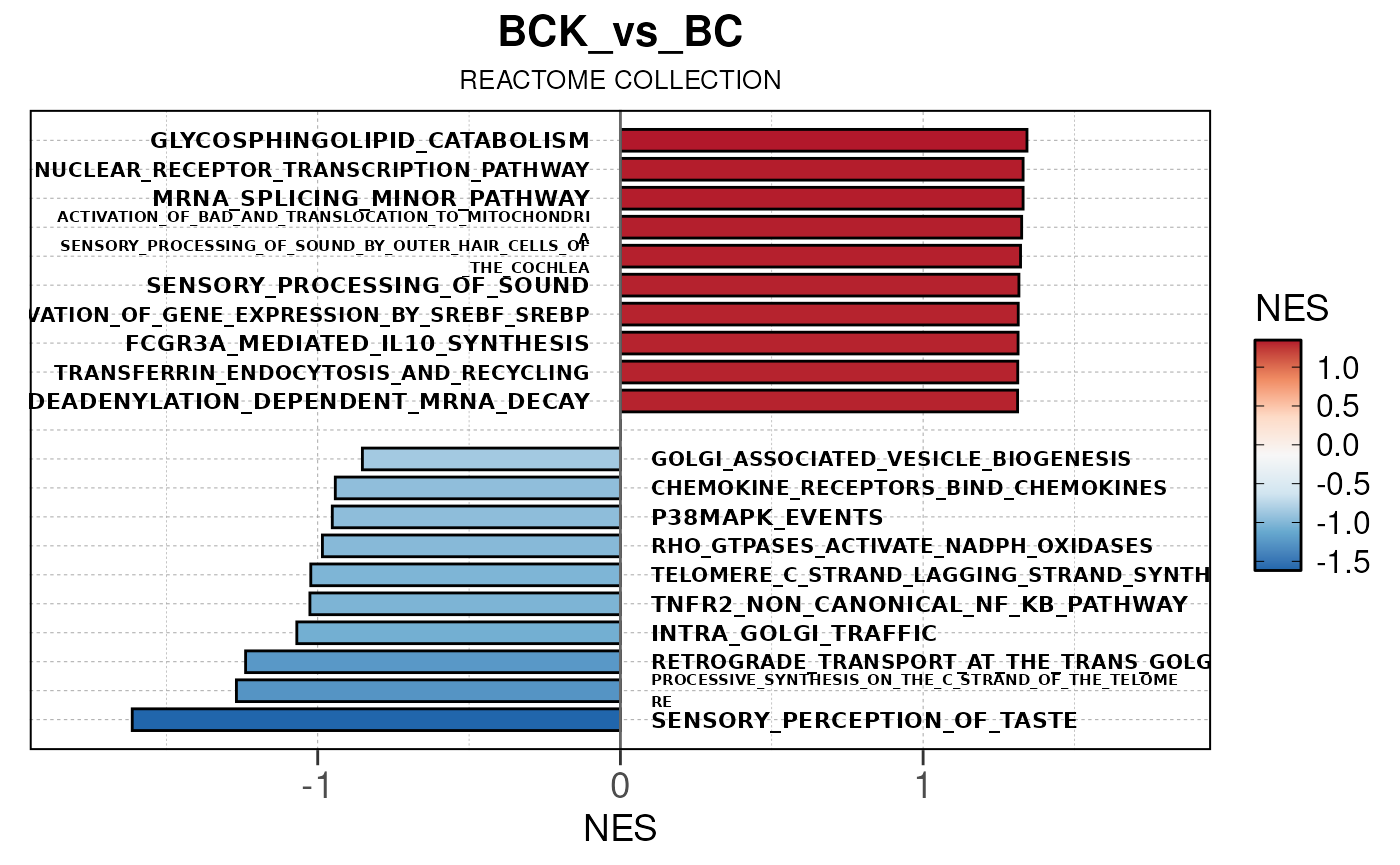
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
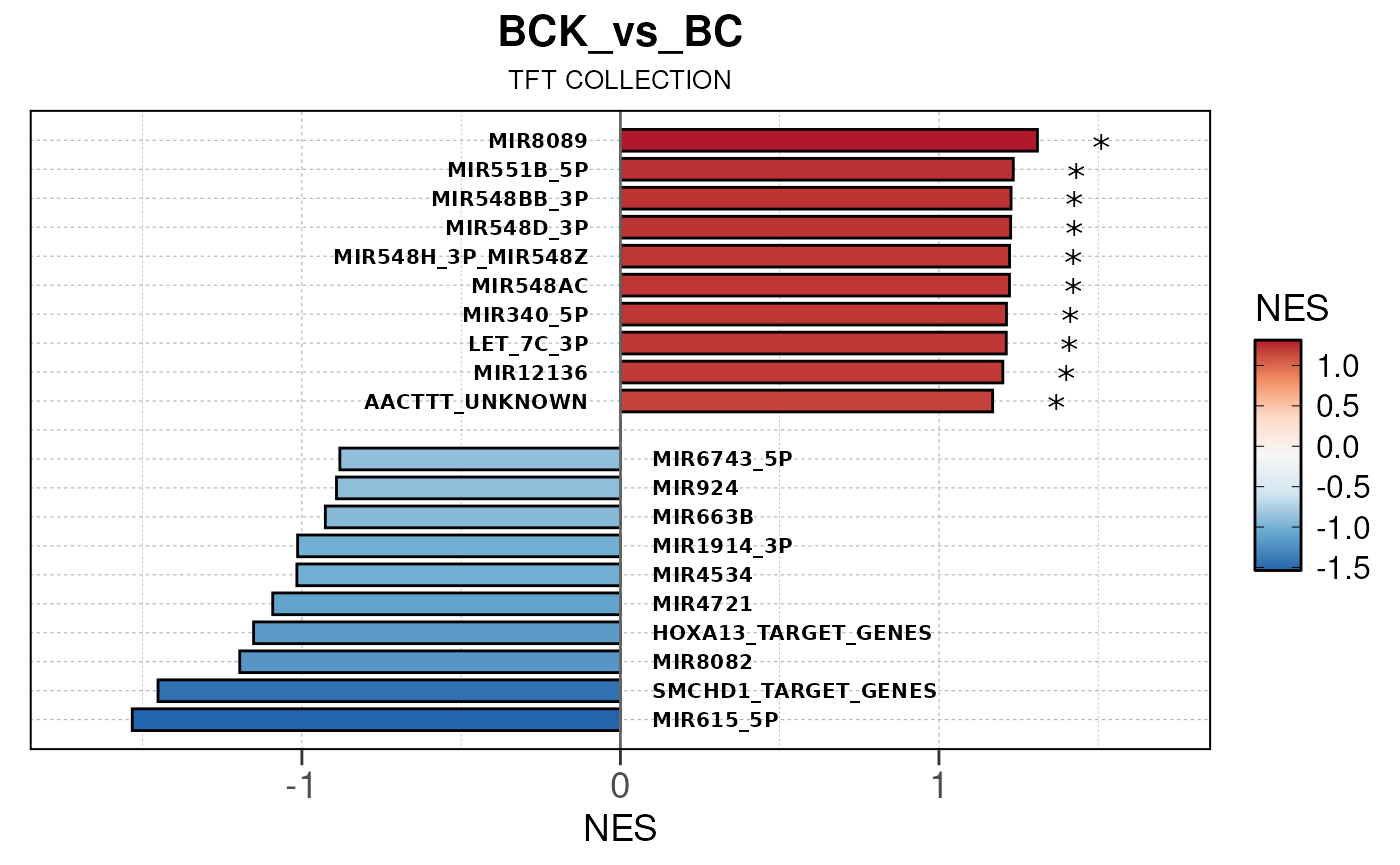
## Running PAIRWISE enrichment map pipeline for TSS PEAKS...
## Formatting for enrichmentmap...
## Writing class files for PAIRWISE comparisons...
## Running ONE-TO-ALL differential expression analysis for TSS PEAKS...
## Running ONE-TO-ALL differential expression analysis...
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 13 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 13 unlabeled data points (too many overlaps). Consider increasing max.overlaps
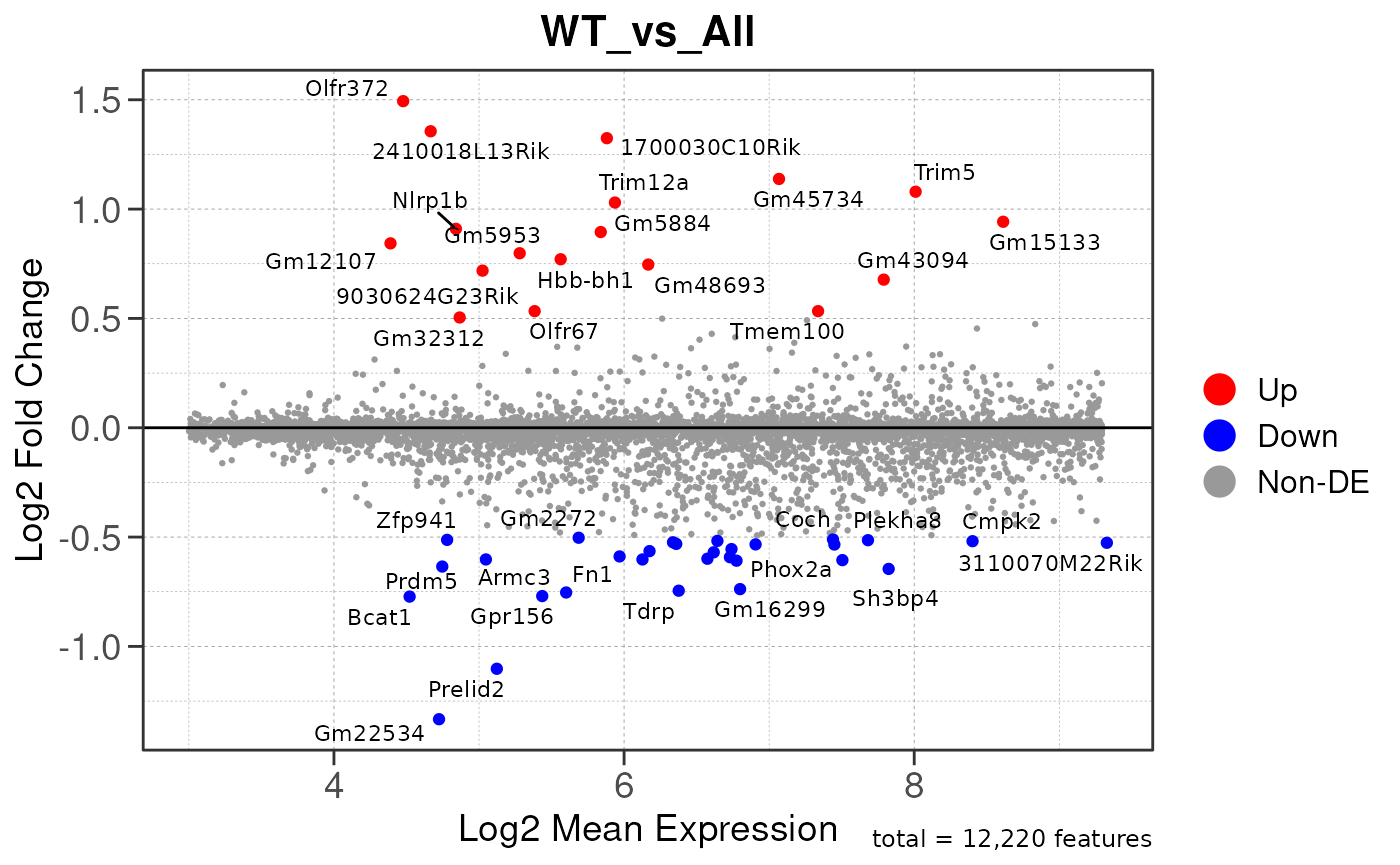
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 12333 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 30 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 12333 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 33 unlabeled data points (too many overlaps). Consider increasing max.overlaps
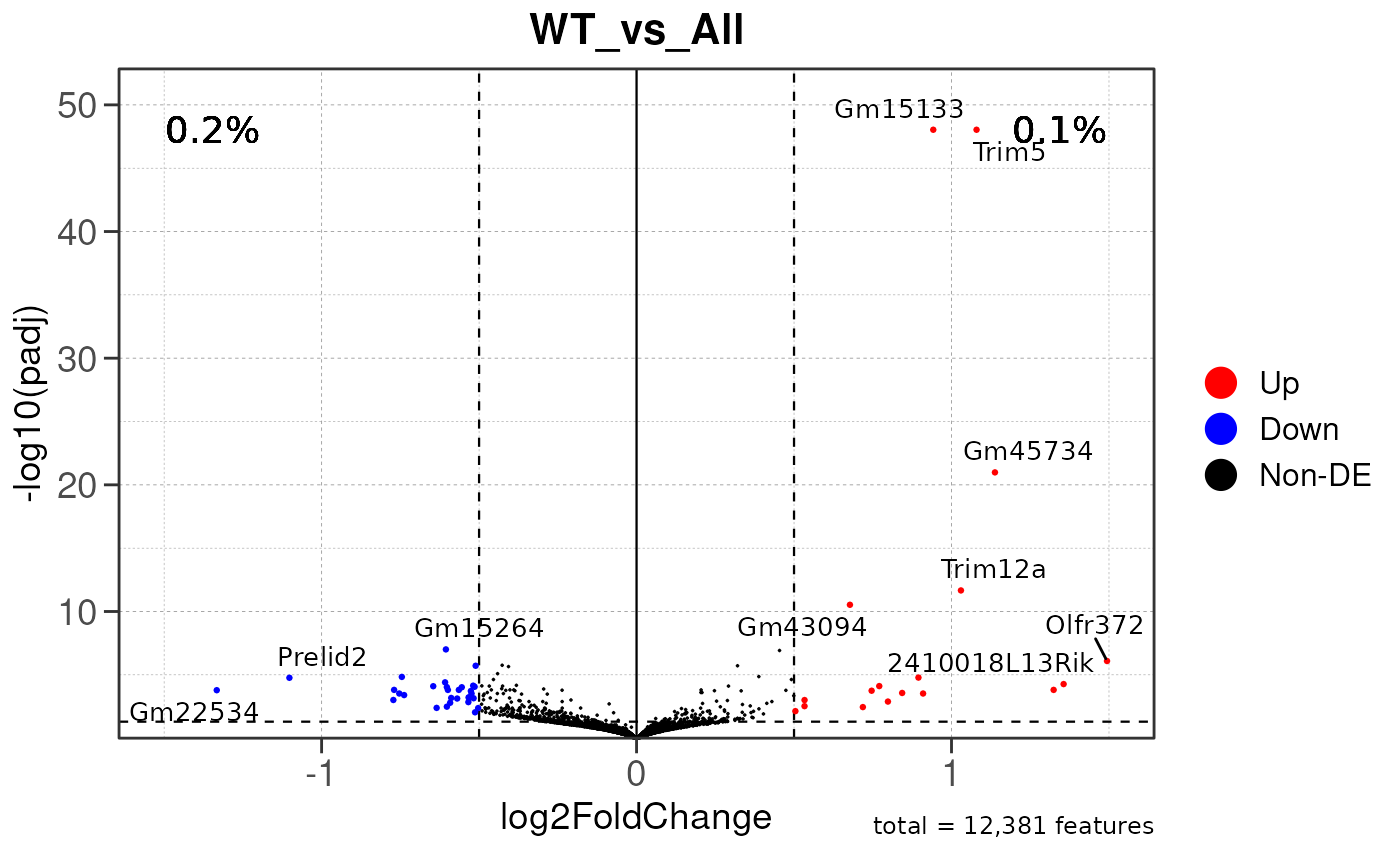
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: ggrepel: 169 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: ggrepel: 171 unlabeled data points (too many overlaps). Consider increasing max.overlaps
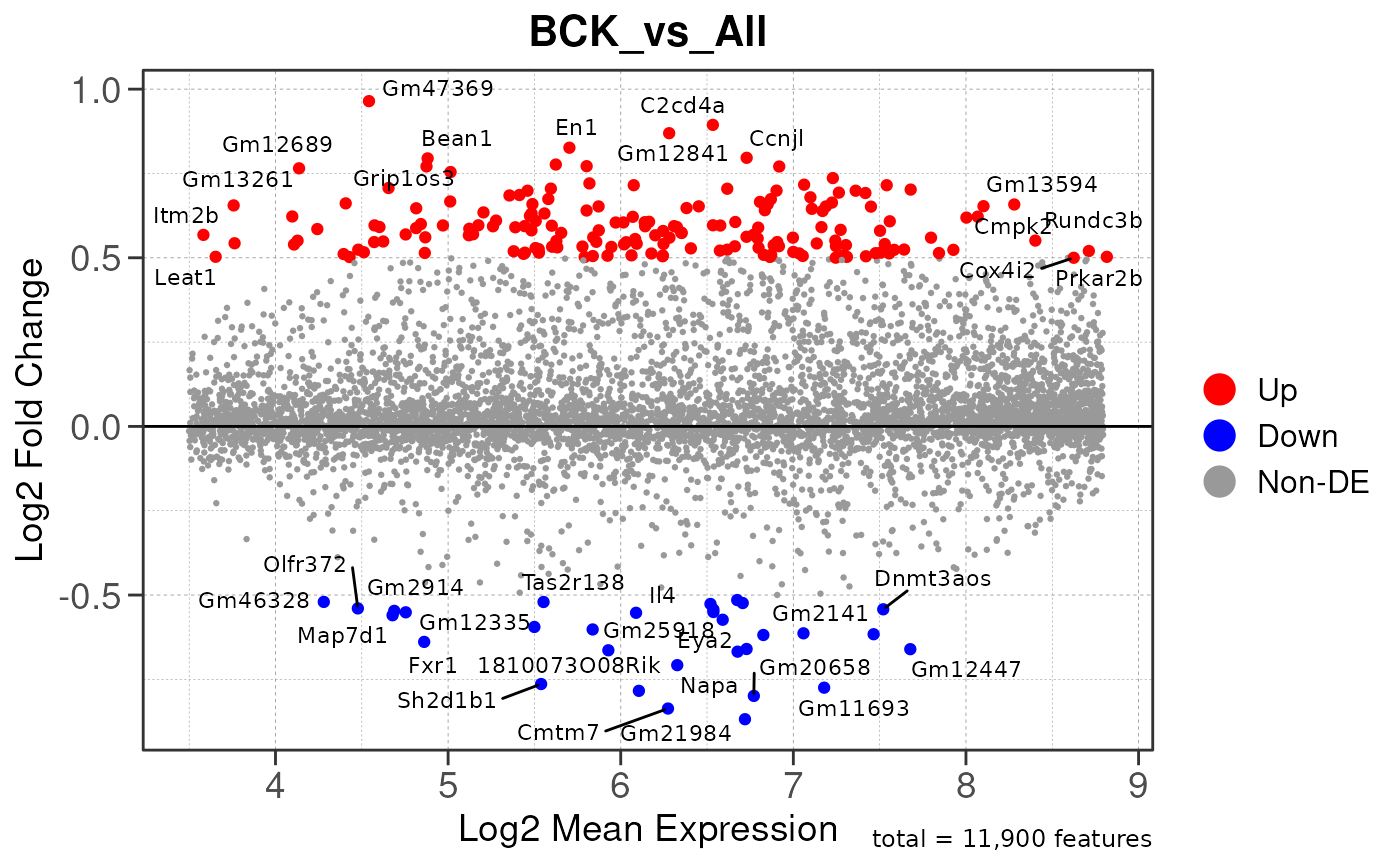
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: Removed 11693 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 22 unlabeled data points (too many overlaps). Consider increasing max.overlaps
## WARNING: Removed 11693 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: ggrepel: 26 unlabeled data points (too many overlaps). Consider increasing max.overlaps
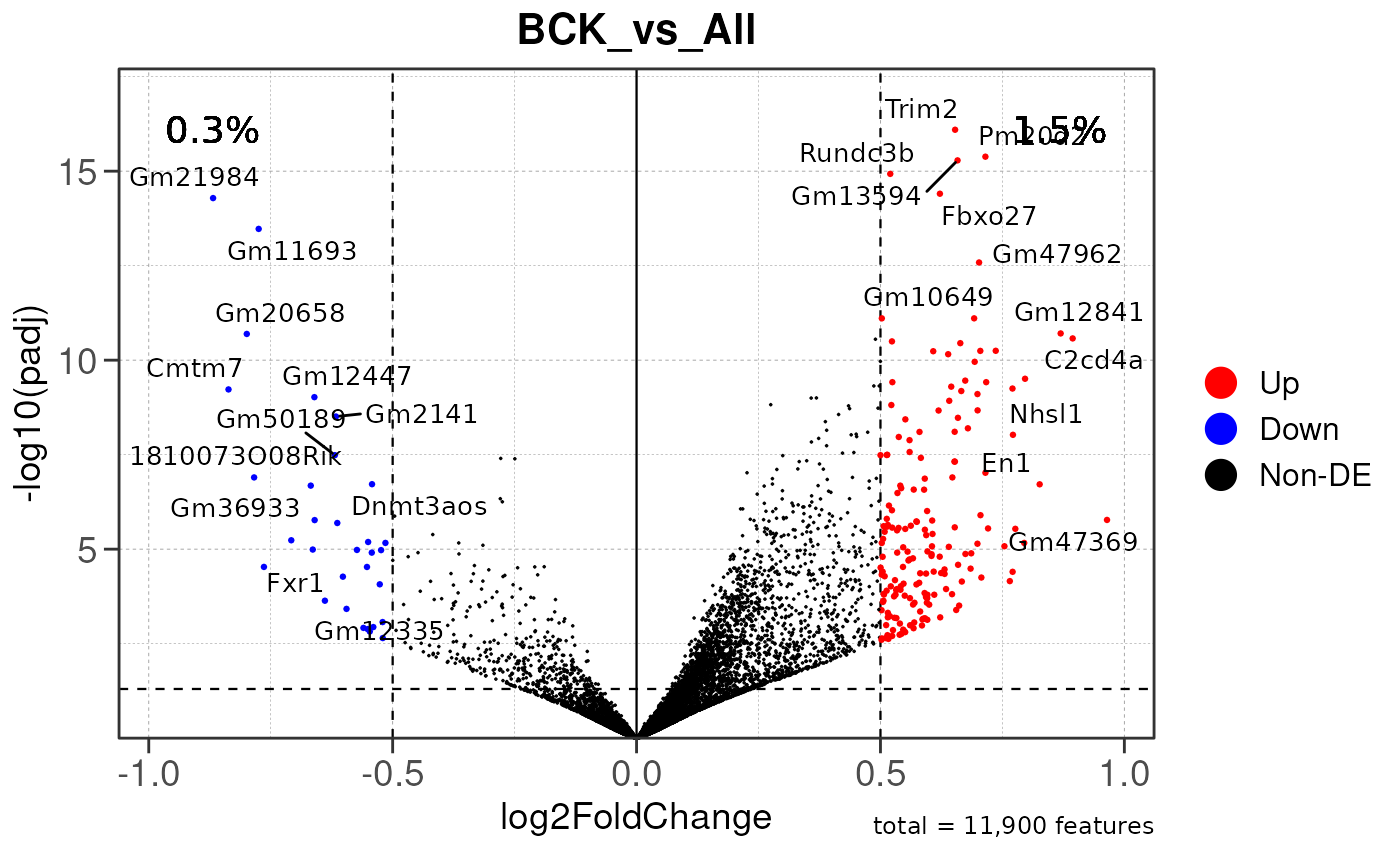
## using pre-existing size factors
## estimating dispersions
## gene-wise dispersion estimates
## mean-dispersion relationship
## final dispersion estimates
## fitting model and testing
## using 'ashr' for LFC shrinkage. If used in published research, please cite:
## Stephens, M. (2016) False discovery rates: a new deal. Biostatistics, 18:2.
## https://doi.org/10.1093/biostatistics/kxw041
## Combining row metadata to results...
## Removed 1 rows with NA values or invalid gene symbols
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: No shared levels found between `names(values)` of the manual scale and the
## data's colour values.
## WARNING: No shared levels found between `names(values)` of the manual scale and the
## data's colour values.

## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## WARNING: no non-missing arguments to max; returning -Inf
## WARNING: NaNs produced
## WARNING: Removed 12381 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 12381 rows containing missing values or values outside the scale range
## (`geom_text()`).
## Running ONE-TO-ALL gene set enrichment pipeline for TSS PEAKS...
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
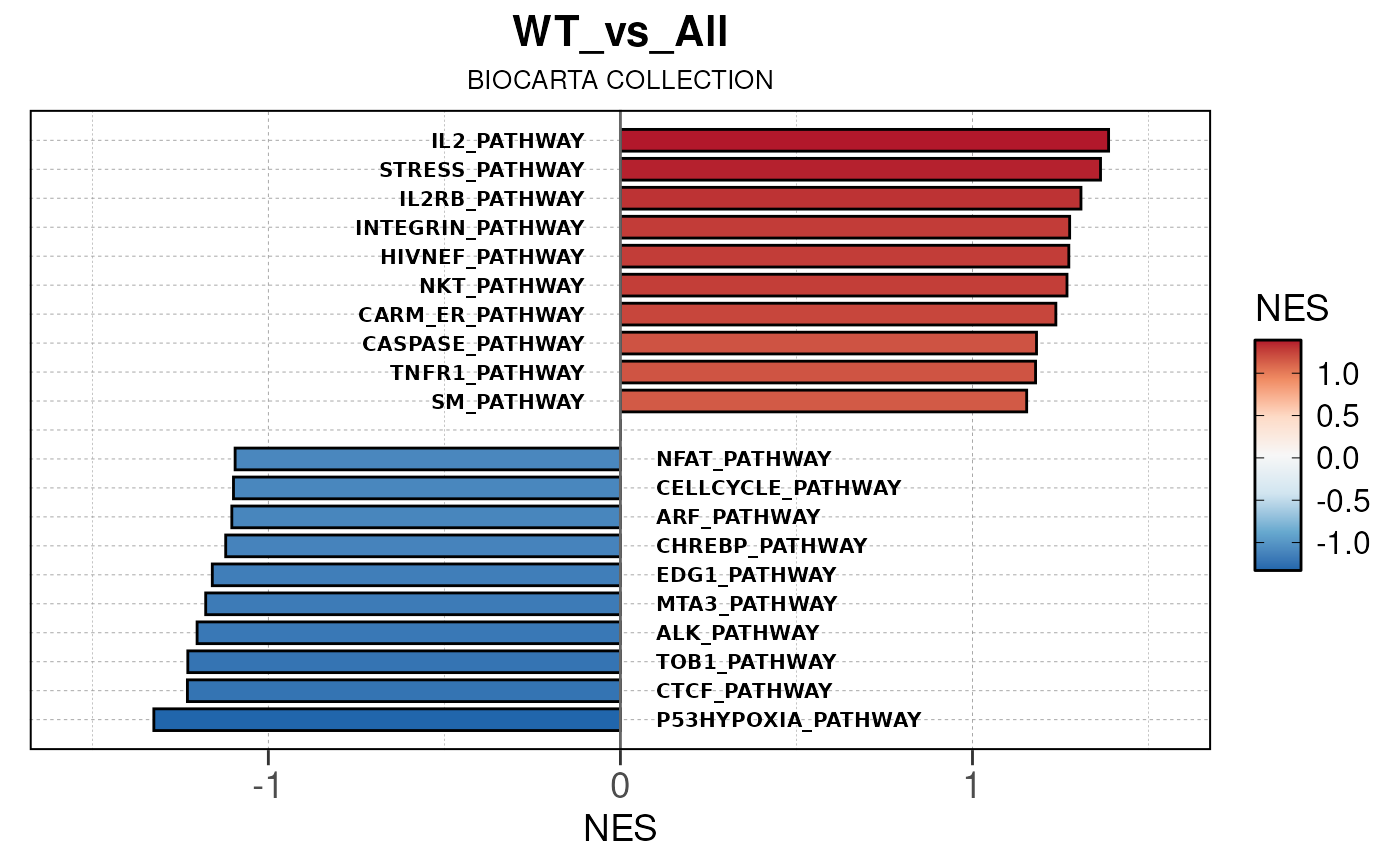
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
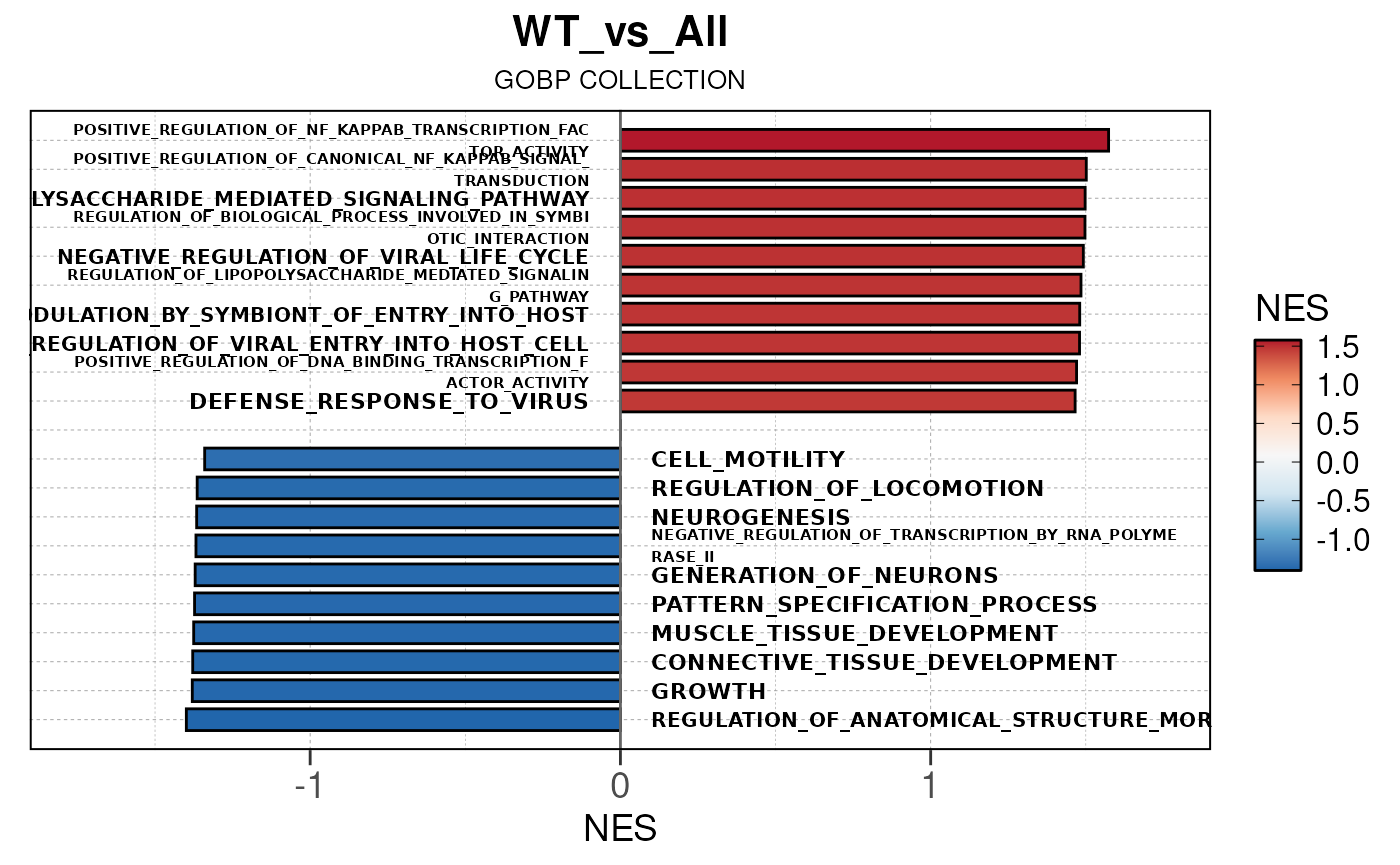
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
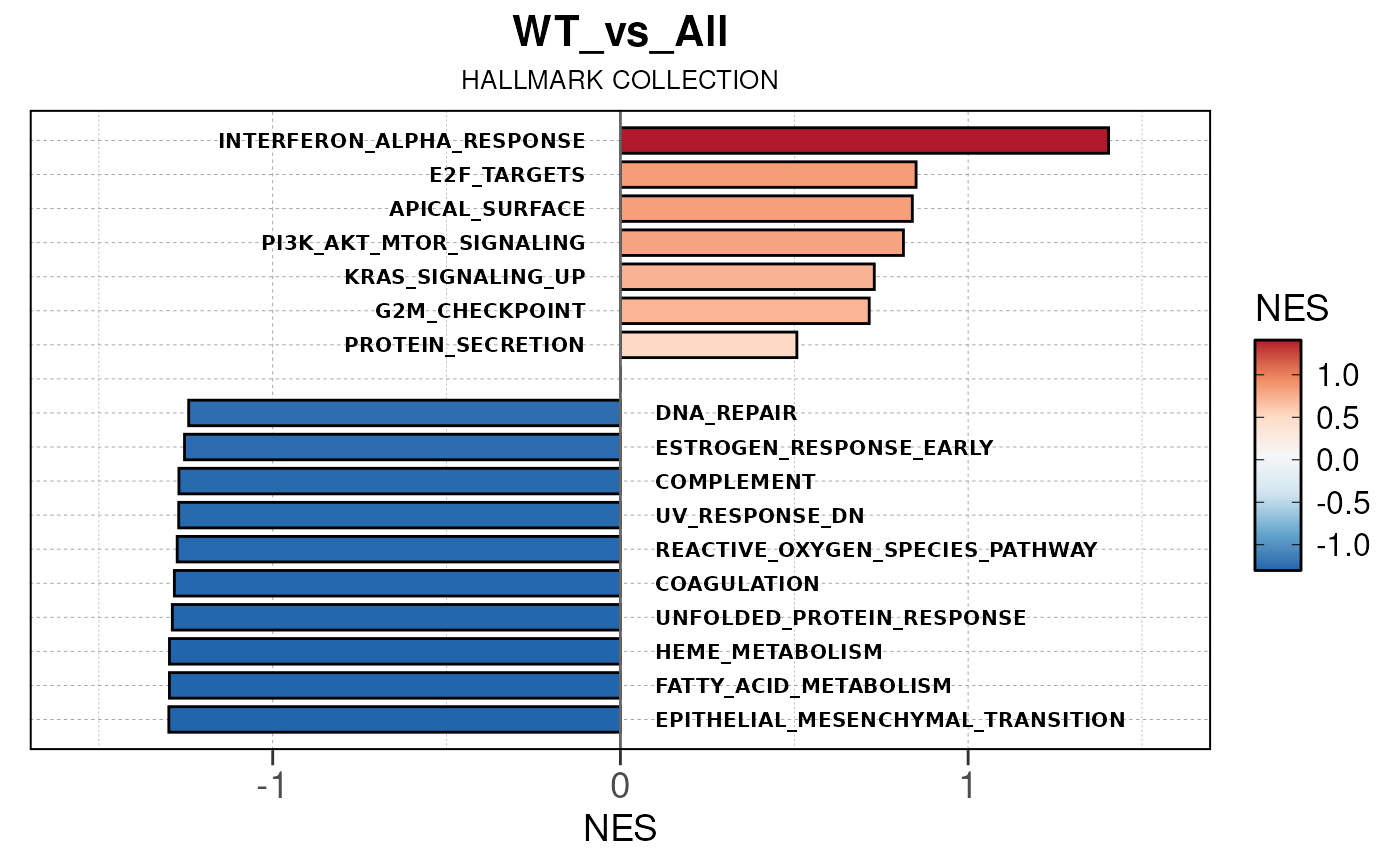
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
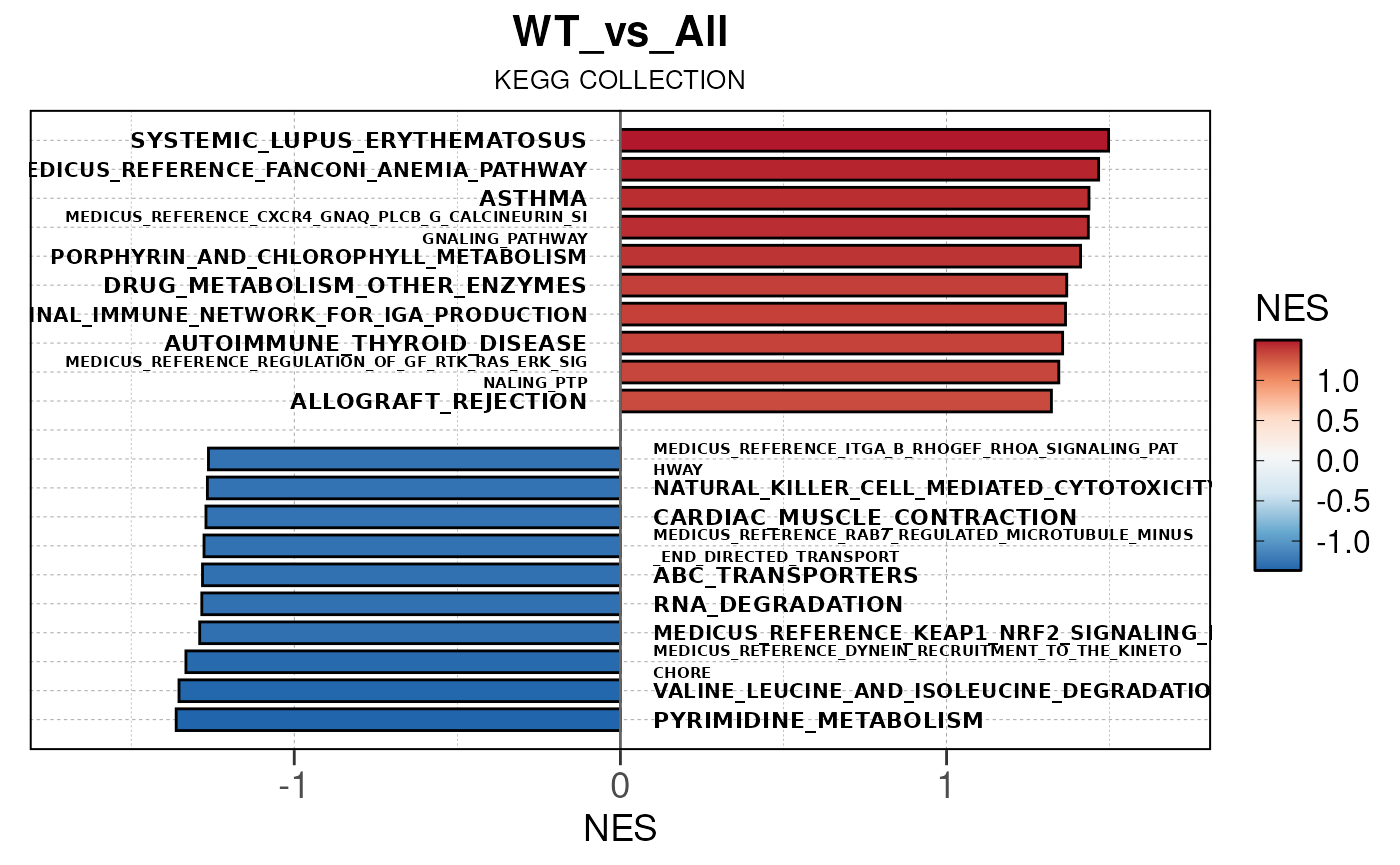
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 9 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 9 rows containing missing values or values outside the scale range
## (`geom_text()`).
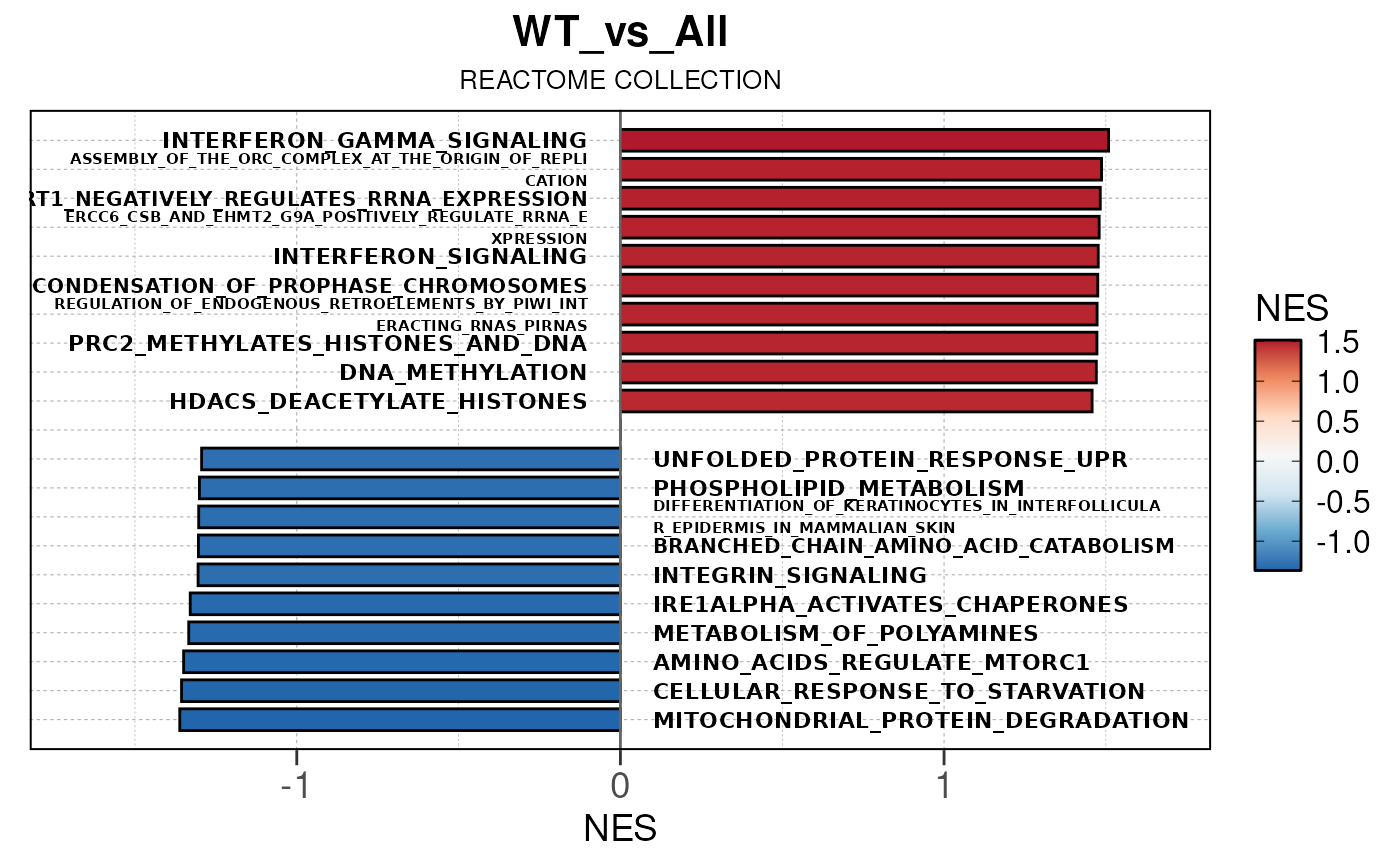
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 5 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 5 rows containing missing values or values outside the scale range
## (`geom_text()`).
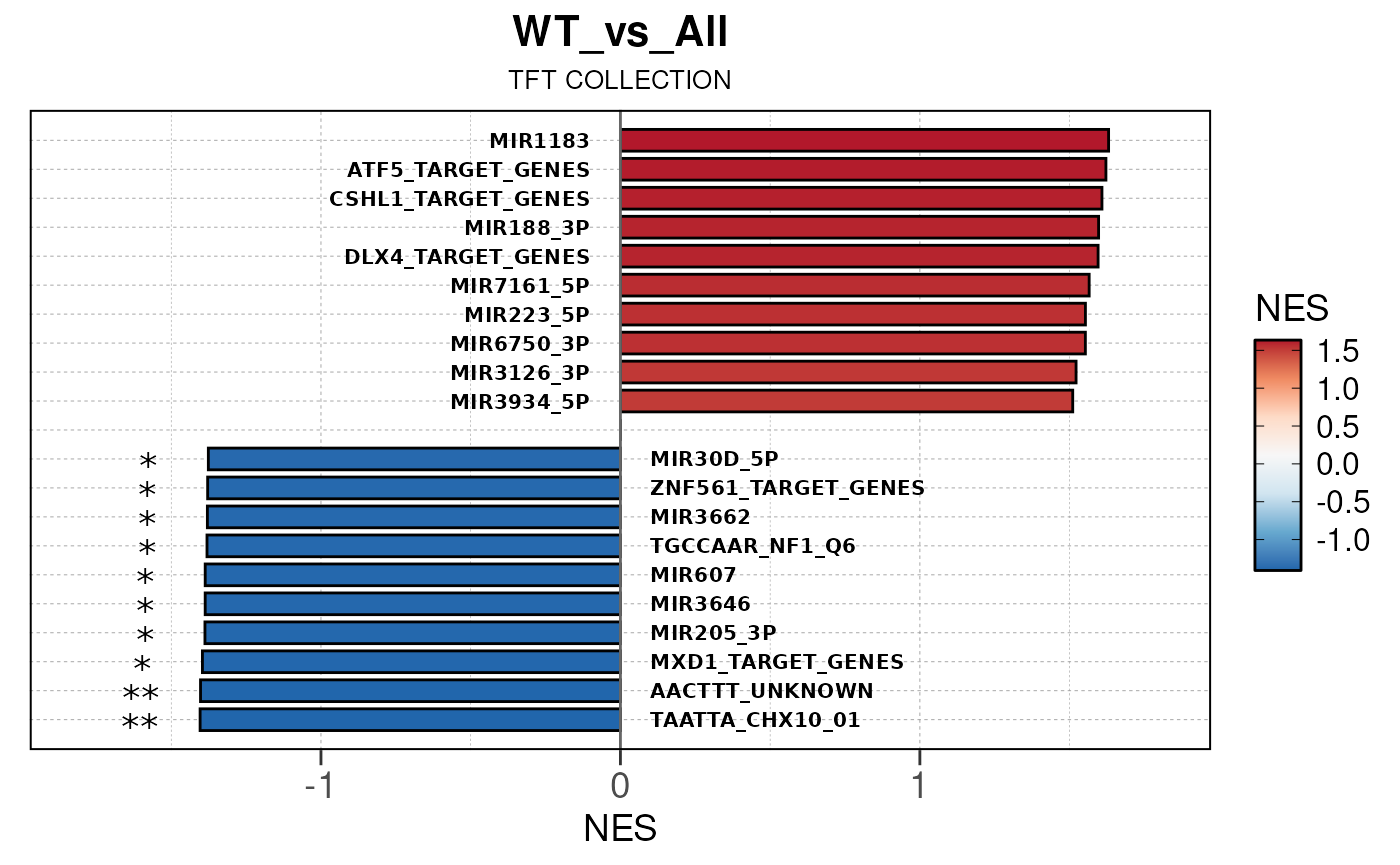
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
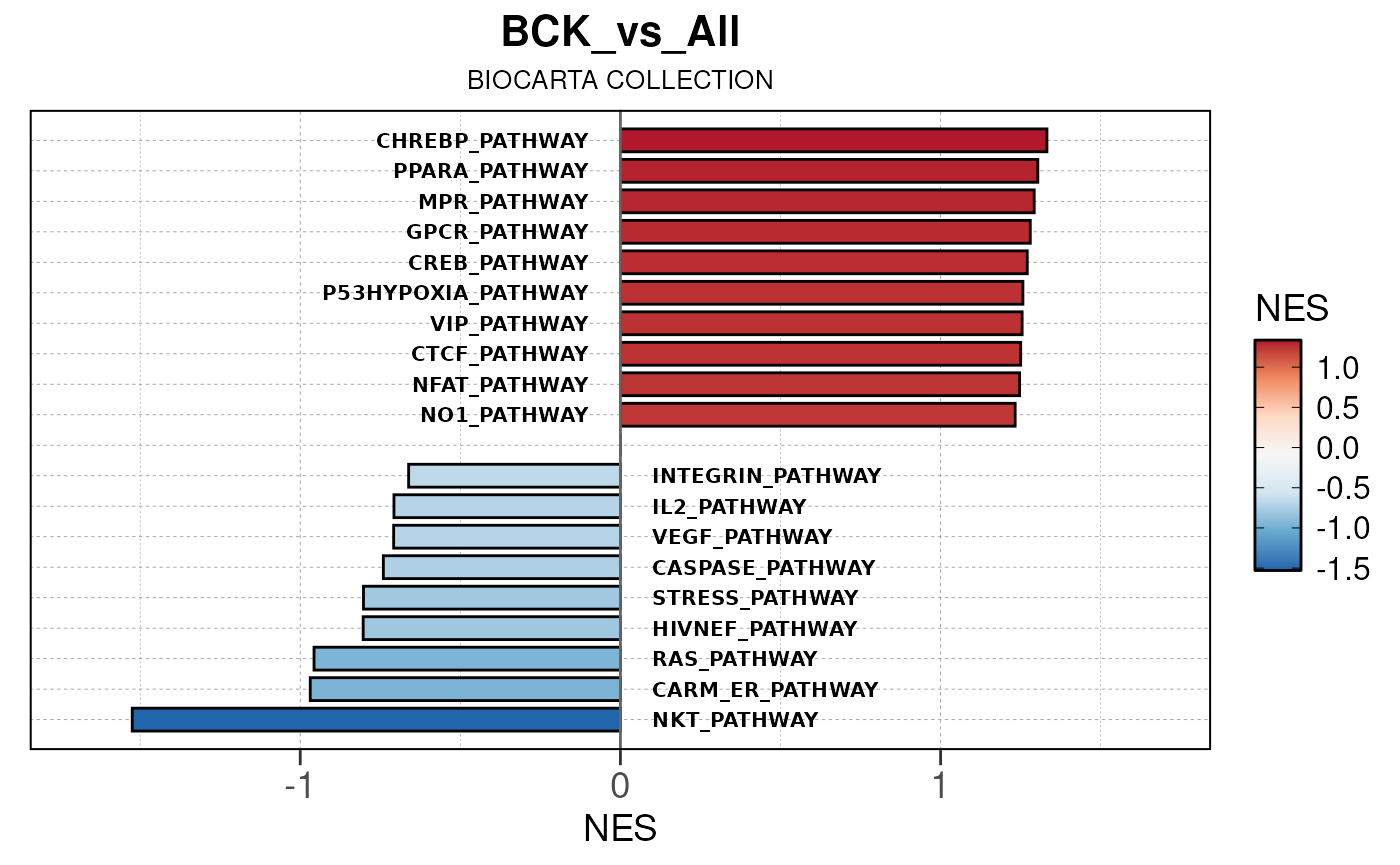
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
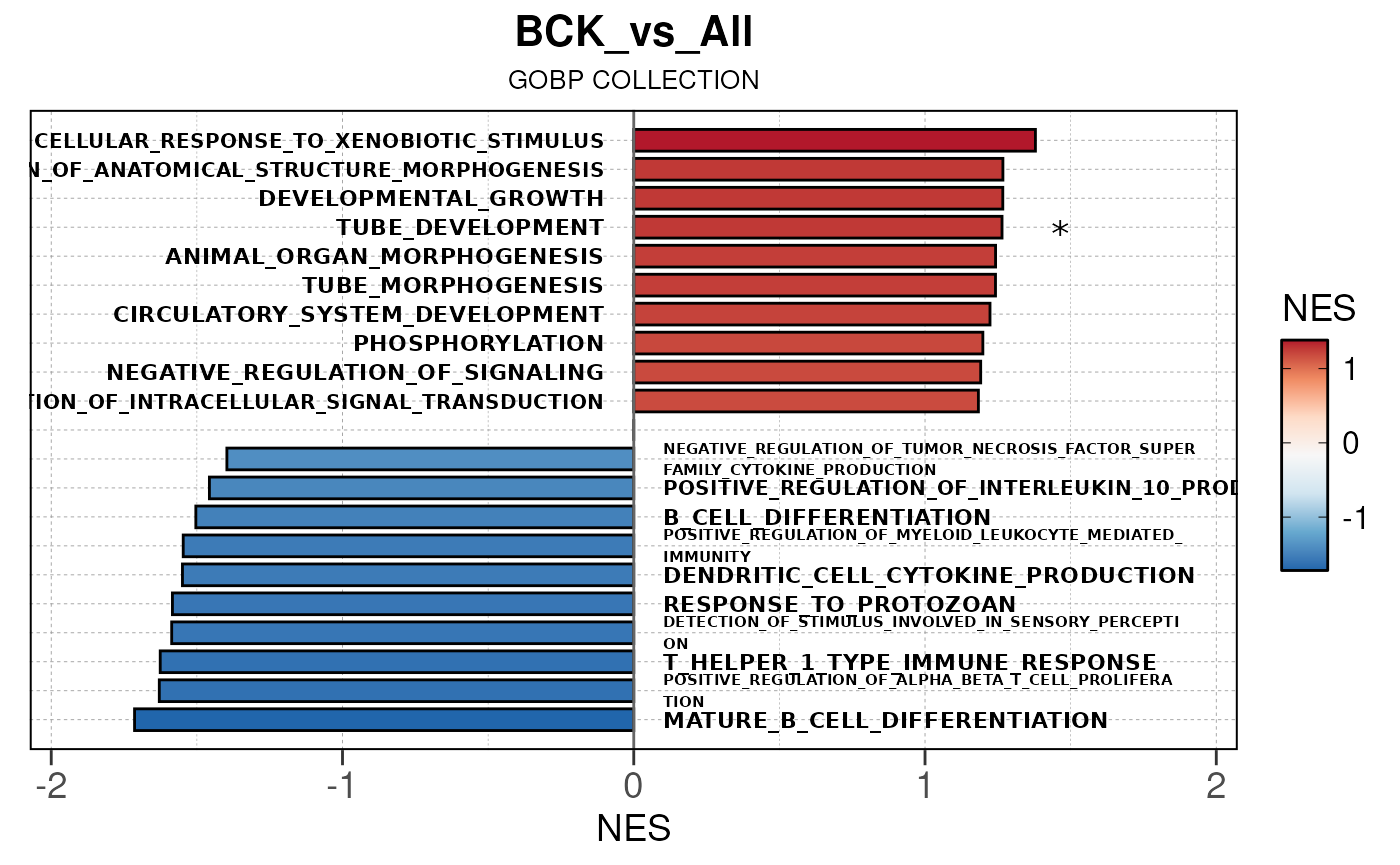
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 6 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 6 rows containing missing values or values outside the scale range
## (`geom_text()`).
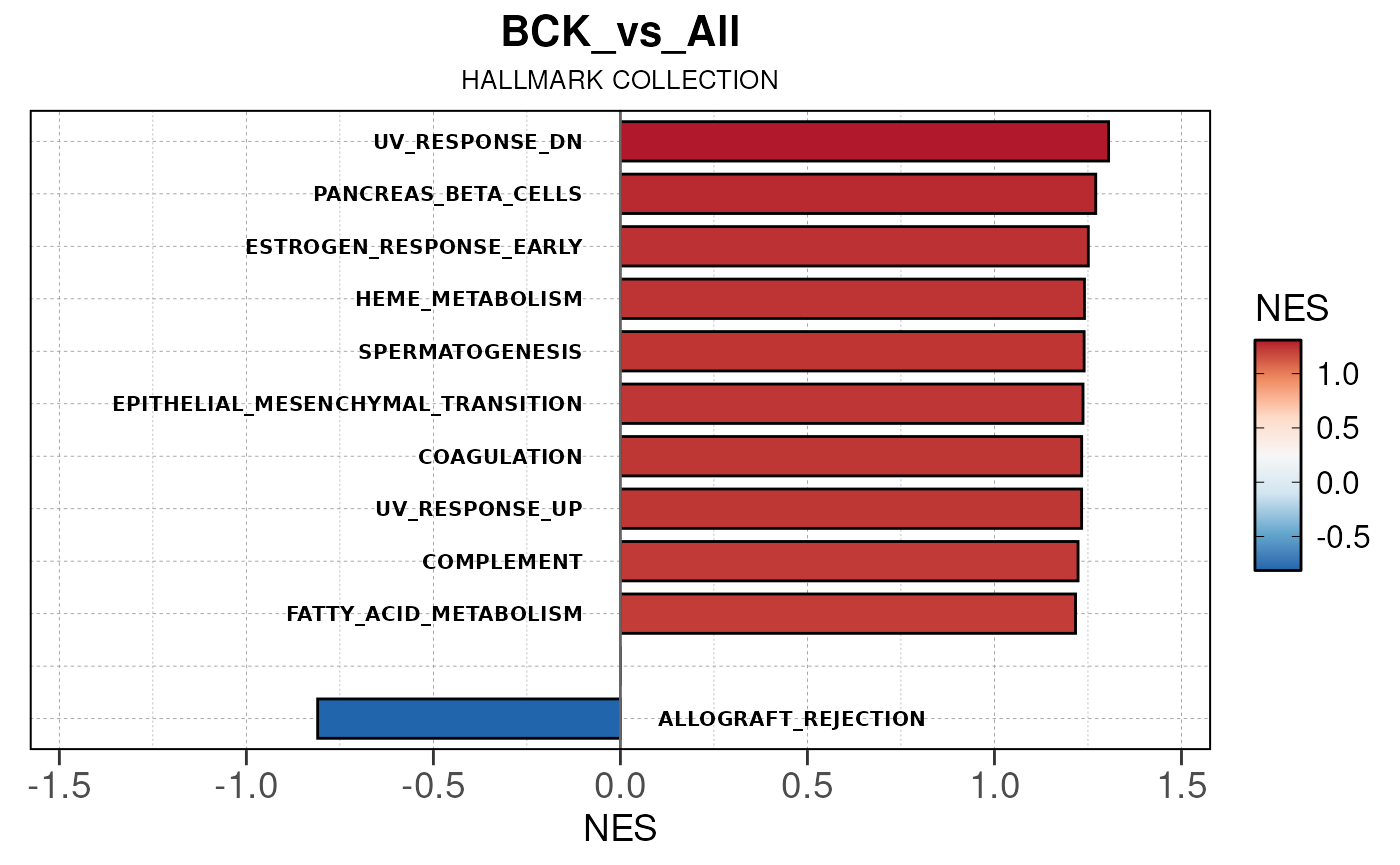
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
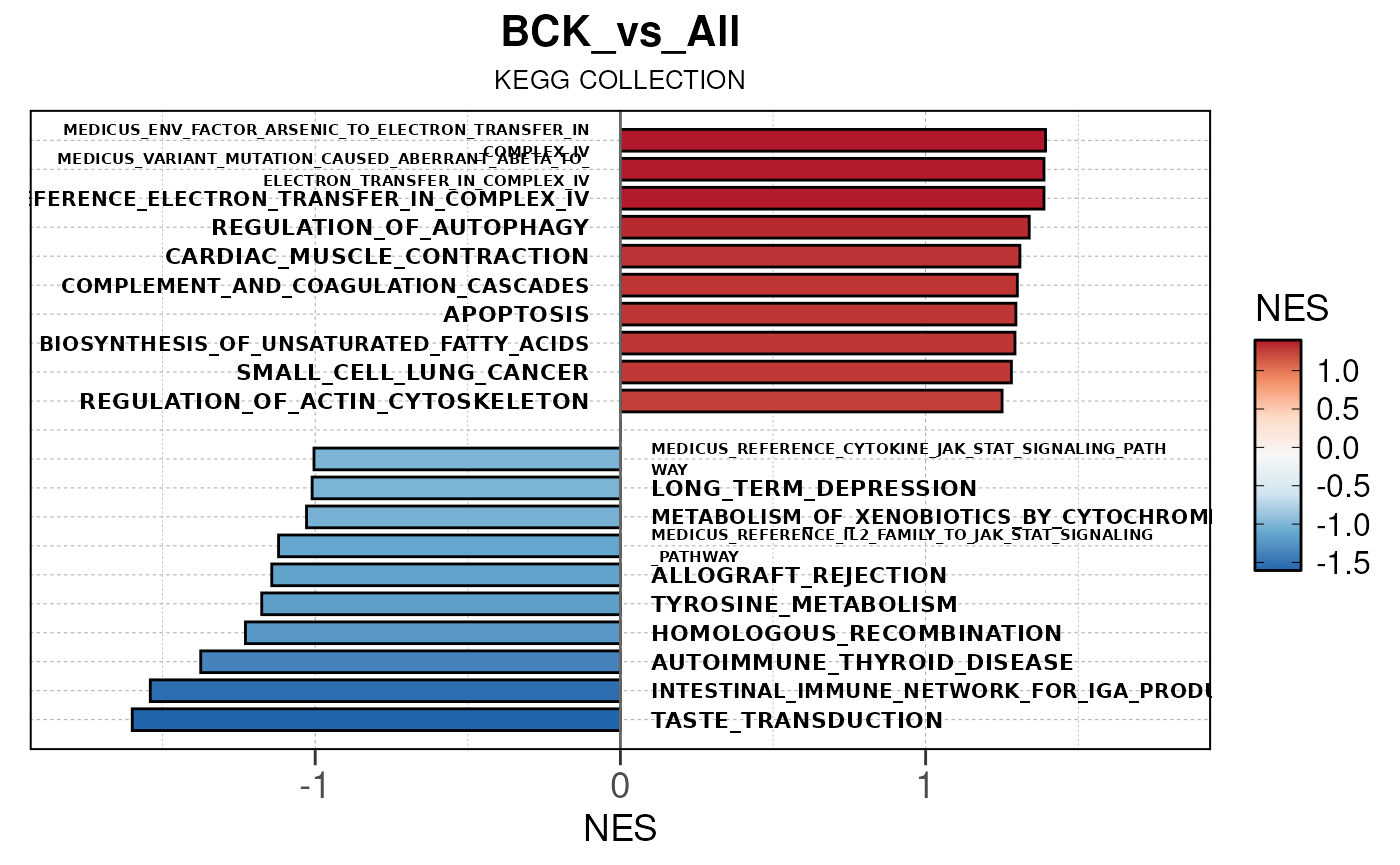
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
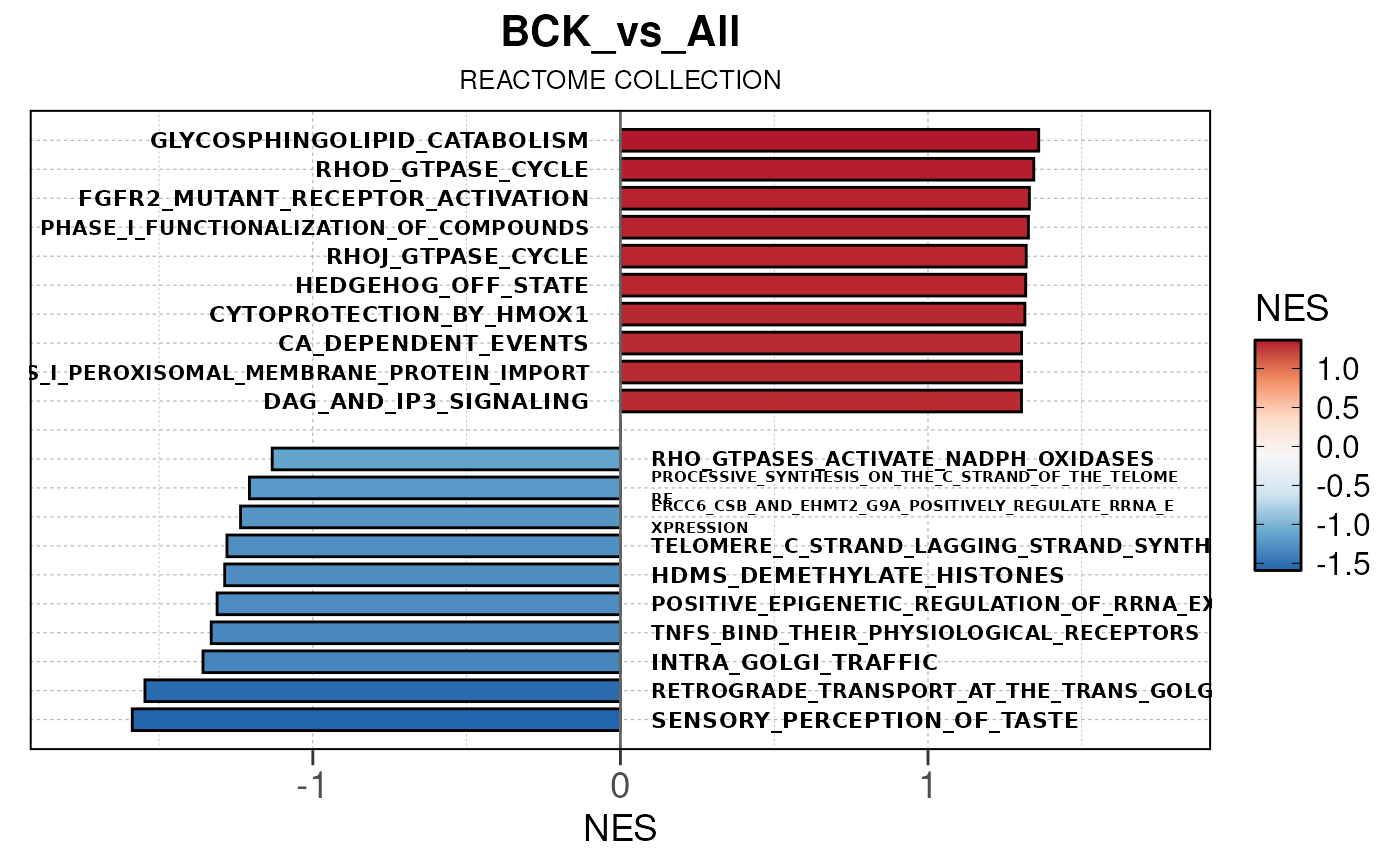
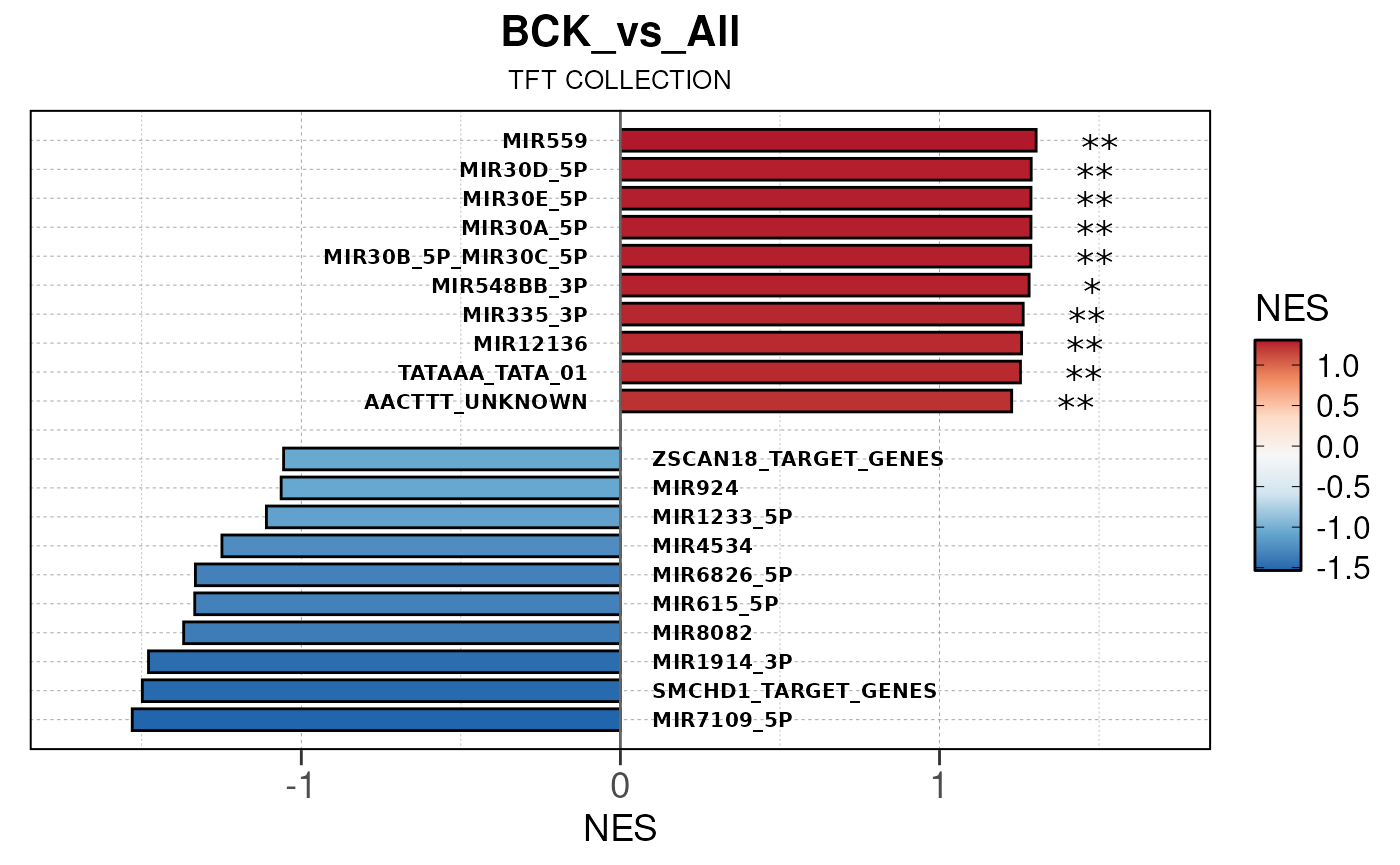
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 2 rows containing missing values or values outside the scale range
## (`geom_text()`).
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## Converting baseMean to numeric
## Converting log2FoldChange to numeric
## Converting padj to numeric
## Converting rank to numeric
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## WARNING: There were 7 pathways for which P-values were not calculated properly due to unbalanced (positive and negative) gene-level statistic values. For such pathways pval, padj, NES, log2err are set to NA. You can try to increase the value of the argument nPermSimple (for example set it nPermSimple = 10000)
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## WARNING: There were 2 pathways for which P-values were not calculated properly due to unbalanced (positive and negative) gene-level statistic values. For such pathways pval, padj, NES, log2err are set to NA. You can try to increase the value of the argument nPermSimple (for example set it nPermSimple = 10000)
## leading edge analysis...
## done...
## using 'fgsea' for GSEA analysis, please cite Korotkevich et al (2019).
## preparing geneSet collections...
## GSEA analysis...
## WARNING: For some of the pathways the P-values were likely overestimated. For such pathways log2err is set to NA.
## WARNING: For some pathways, in reality P-values are less than 1e-10. You can set the `eps` argument to zero for better estimation.
## leading edge analysis...
## done...
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
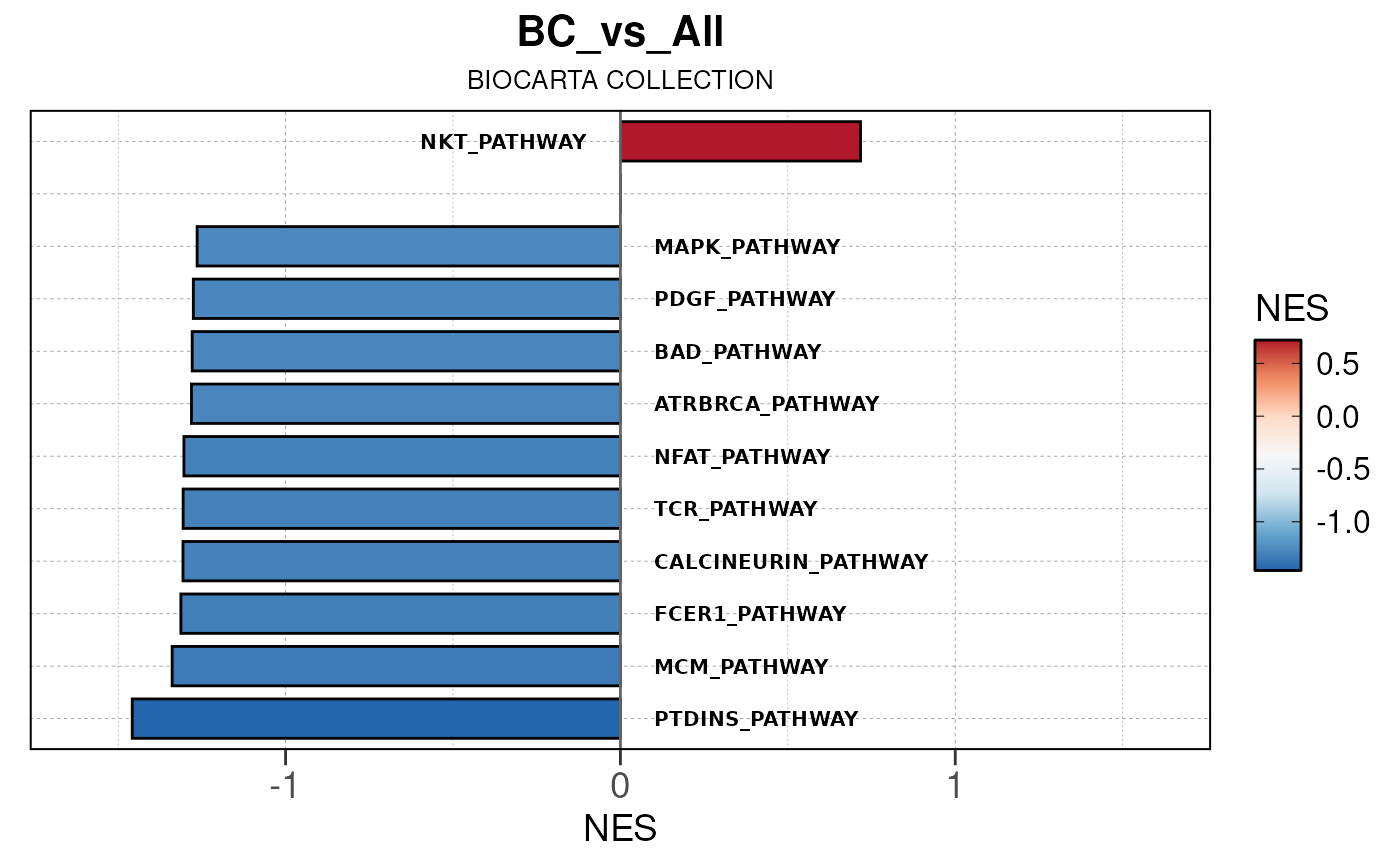
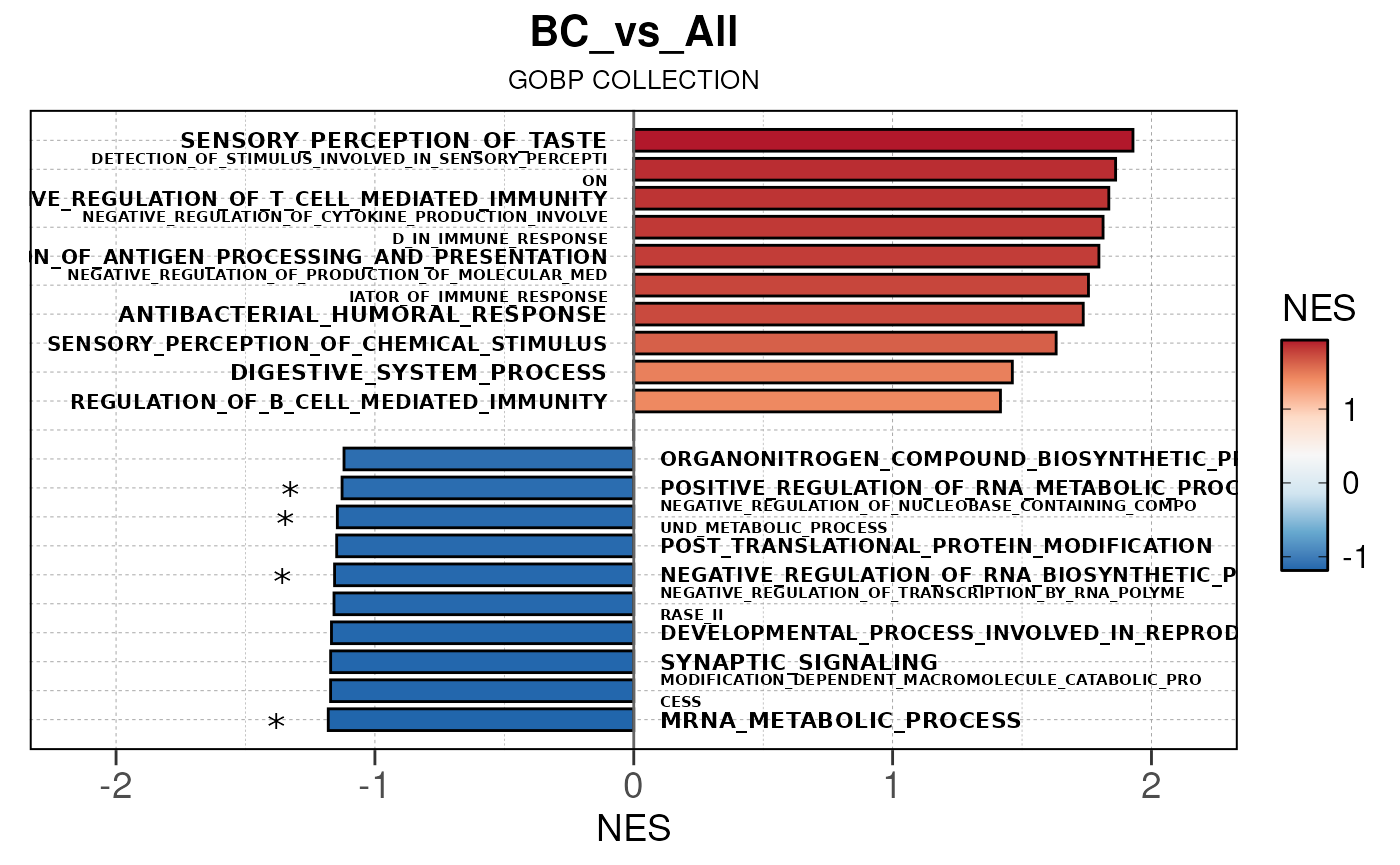
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 6 rows containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 6 rows containing missing values or values outside the scale range
## (`geom_text()`).
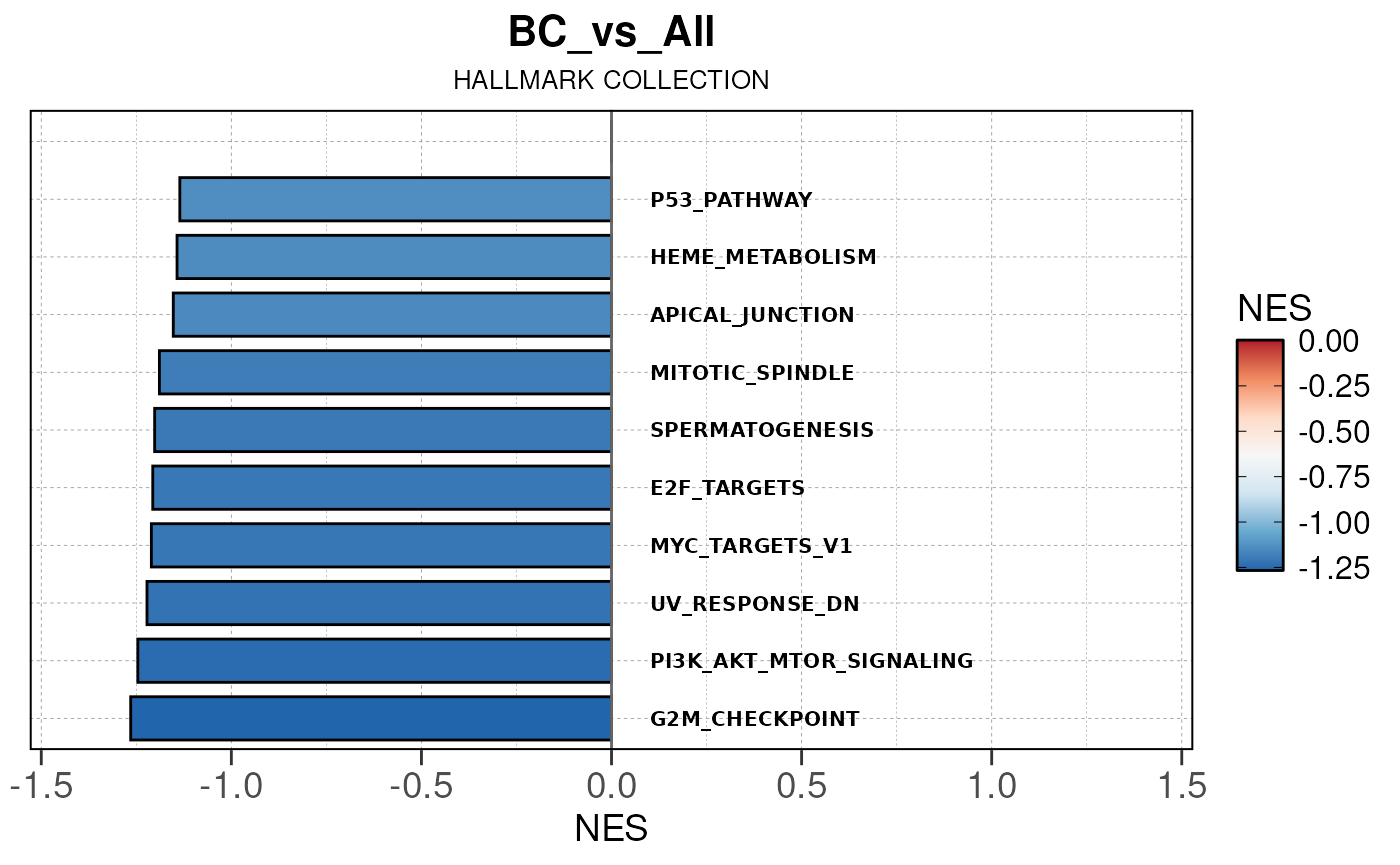
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
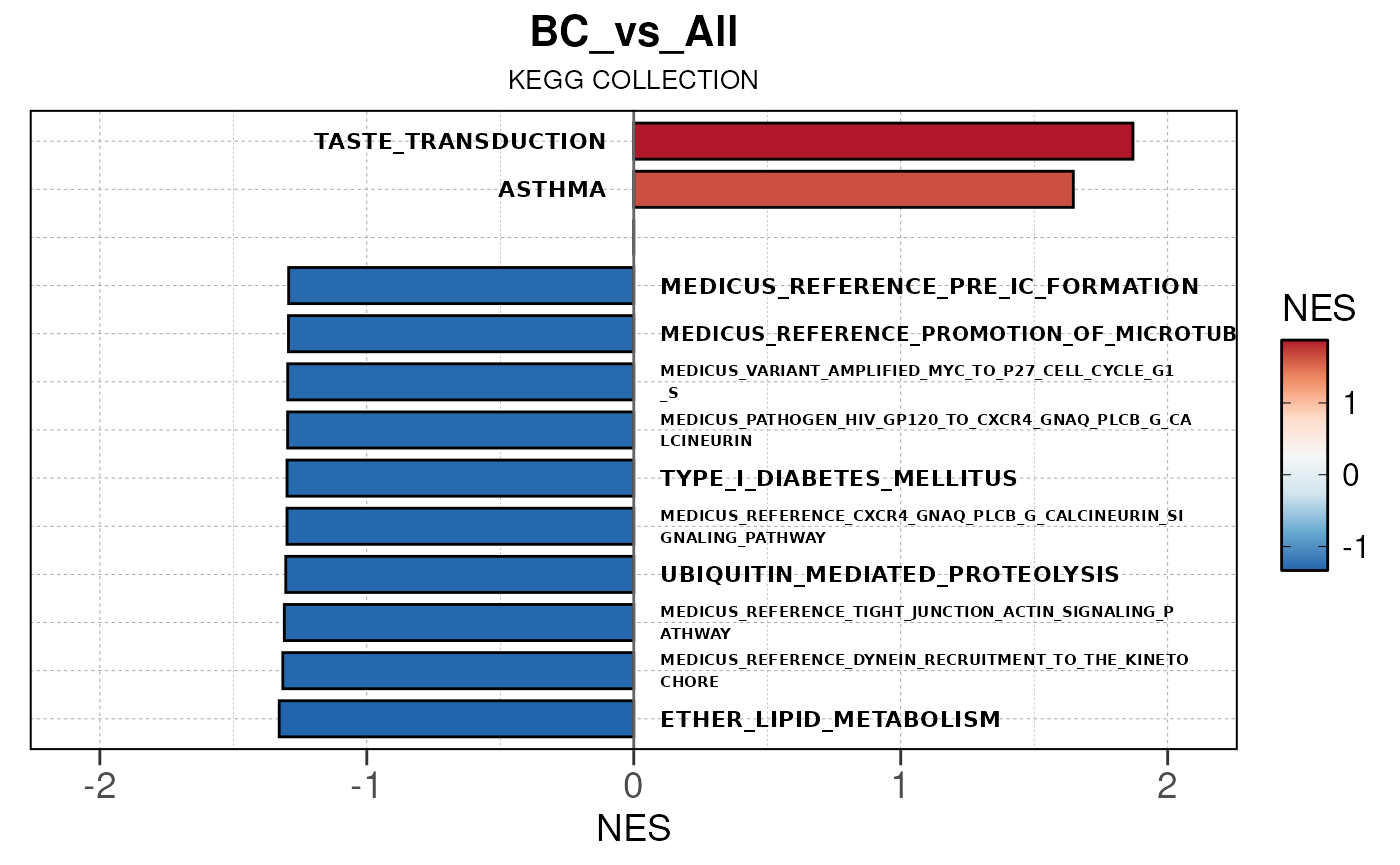
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
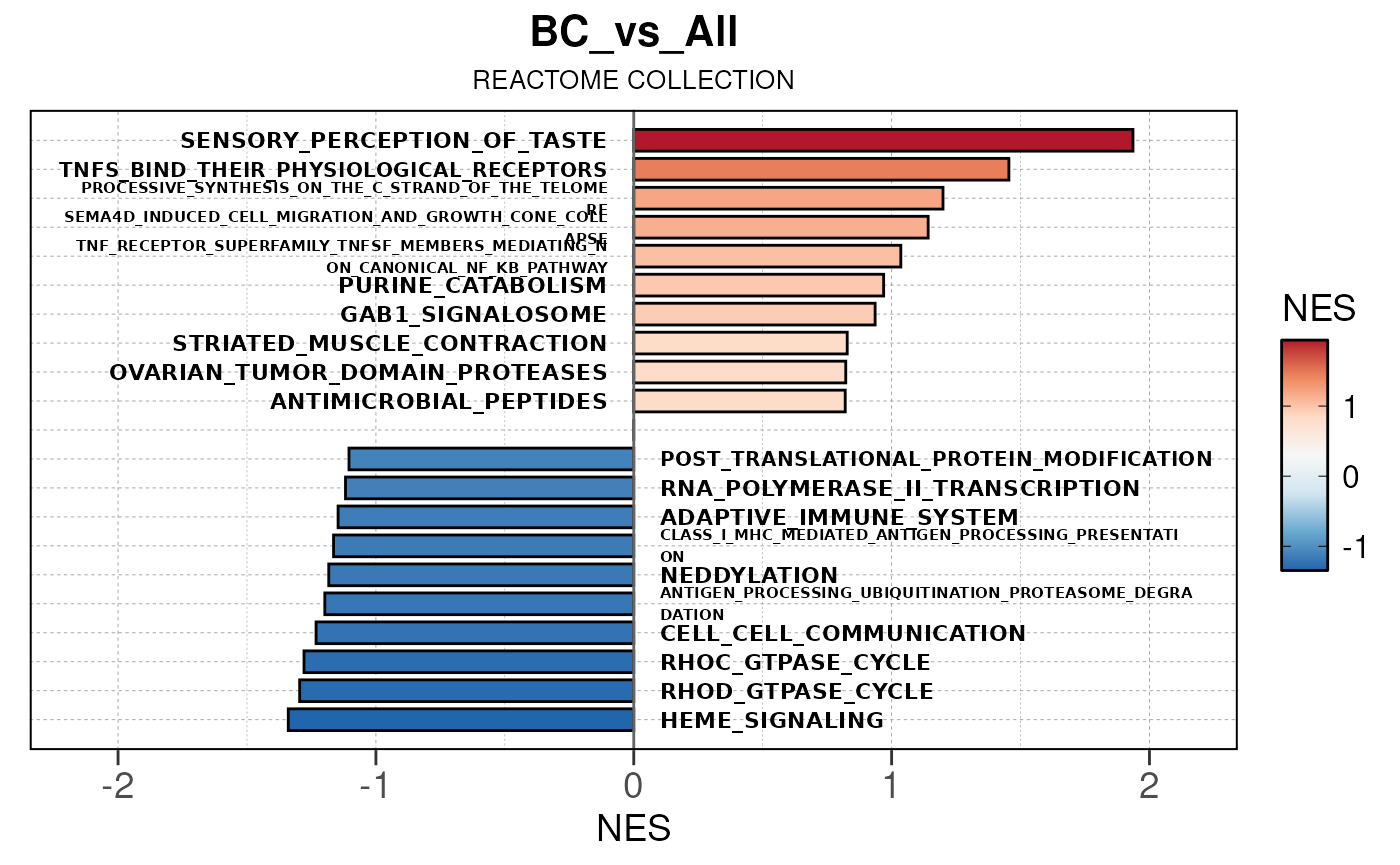
## Converting NES to numeric
## Converting pvalue to numeric
## Converting qvalue to numeric
## WARNING: Ignoring unknown aesthetics: stroke
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
## WARNING: Removed 1 row containing missing values or values outside the scale range
## (`geom_text()`).
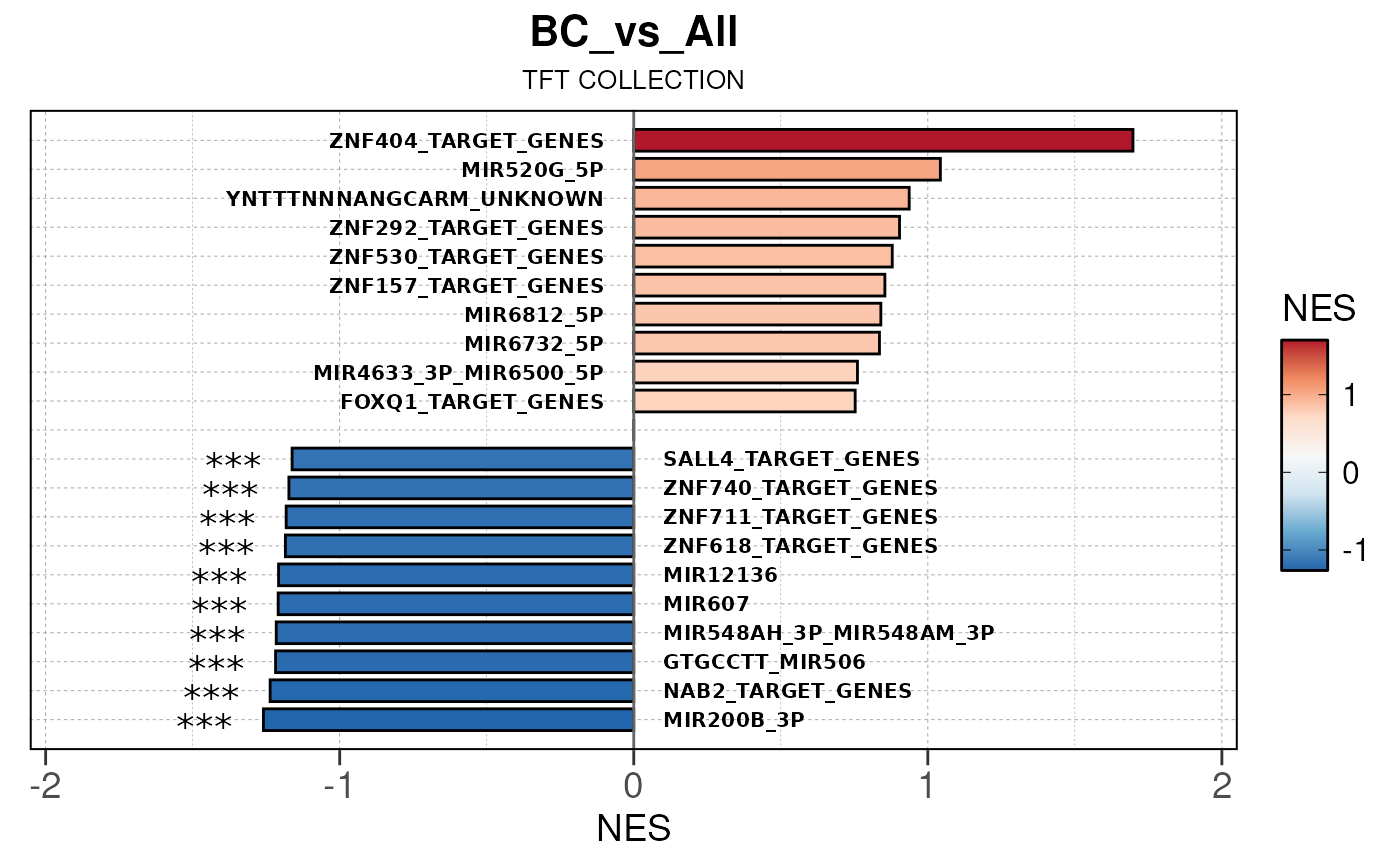
## Running ONE-TO-ALL enrichment map pipeline for TSS PEAKS...
## Formatting for enrichmentmap...
## Writing class files for ONE-TO-ALL comparisons...
## ATACseq pipeline complete; returning DESeq2 object...
## class: DESeqDataSet
## dim: 69431 13
## metadata(1): version
## assays(2): counts vst
## rownames(69431): Interval_1 Interval_2 ... Interval_74799
## Interval_74821
## rowData names(20): peaks Chr ... gene TSS
## colnames(13): WT_REP4 BCK_REP5 ... BC_REP2 BC_REP1
## colData names(4): sample Group1 Group2 sizeFactor